Inno-Docking
1. Inno-Docking 概述
Docking 作为基于结构的药物设计中的常规技术,已被广泛用于识别化合物库中的潜在 Hits,它可以帮助我们理解蛋白质与配体之间的结合方式,并估计其结合亲和力。通常情况下,一个对接程序的可靠性主要取决于构象搜索算法的效率及其打分函数的质量,前者专门用于生成配体构象,后者则用于计算蛋白质-配体结合姿势的结合亲和力。
越来越多的研究发现,对接软件中嵌入的打分函数预测的结合亲和力与实验测定的亲和力没有表现出令人满意的相关性,甚至无法有效区分活性和非活性物质。随着计算机技术的快速发展,发现基于机器学习算法的打分函数可以用非线性的方式隐式地学习和捕获蛋白质-配体的结合特征,结果证明无论是评分能力(对结合亲和力进行排序的能力),对接能力(区分原始结合姿势和诱饵结合姿势的能力),还是筛选能力(从诱饵中分辨出活性化合物的能力),基于机器学习算法的打分函数比传统打分函数具有更好的灵活性和更高的性能。
因此,Inno-Docking 模块不仅集成了经典的物理对接程序 AutoDock Vina,还集成了自研的 AI 对接程序 CarsiDock、CarsiDock-Cov 等。相对物理方法,AI 对接程序在对接构象精度上有显著优势。此外,Inno-Docking 还提供了完整的蛋白预处理、配体预处理和自动智能化设置对接参数能力,且在结果页面提供了详细的数据分析能力。
2. 使用说明
在对接的创建任务页面,根据功能可将其划分为两个区:3D 可视化编辑区和对接参数设置区。3D 可视化编辑区,您可以根据自己的习惯改变结构的显示形态,长按鼠标左键可对蛋白进行旋转,长按鼠标右键可对蛋白进行平移,滚动鼠标滚轮可放大缩小蛋白。另外,根据对接的内容,我们把对接分成了三大步,分别为蛋白预处理、配体预处理、设置对接参数。
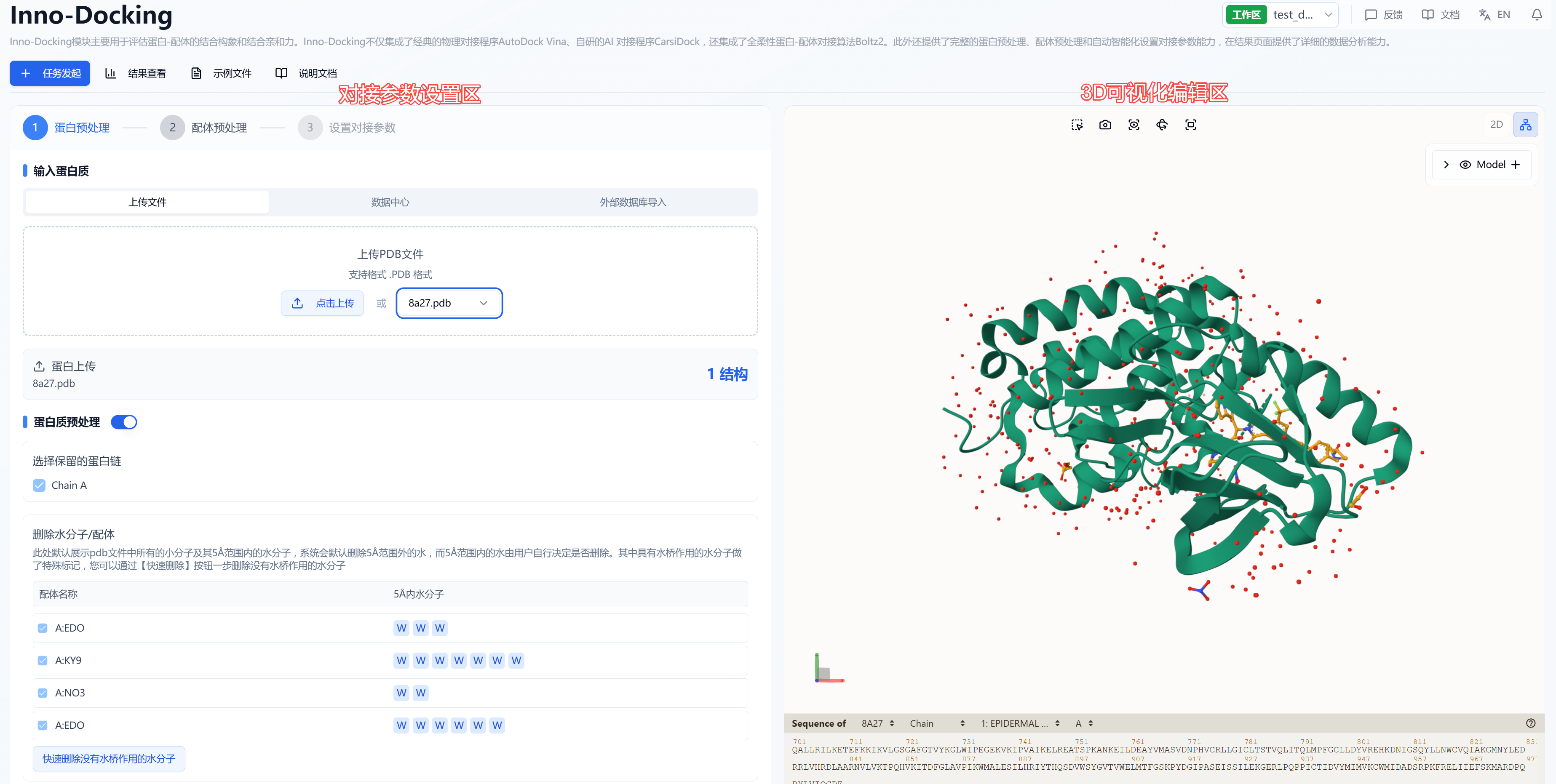
图 1. 对接计算页面介绍
(1) 蛋白预处理
按照常规的蛋白预处理方式,我们提供了相对完善的预处理步骤,包括选择保留的蛋白链、修复错误结构/添加缺失的残基、添加氢原子以及能量最小化等。其面板设置如图 2 所示,页面上默认选择的均为当前最优参数,用户也可根据自己的知识去选择适合的参数。
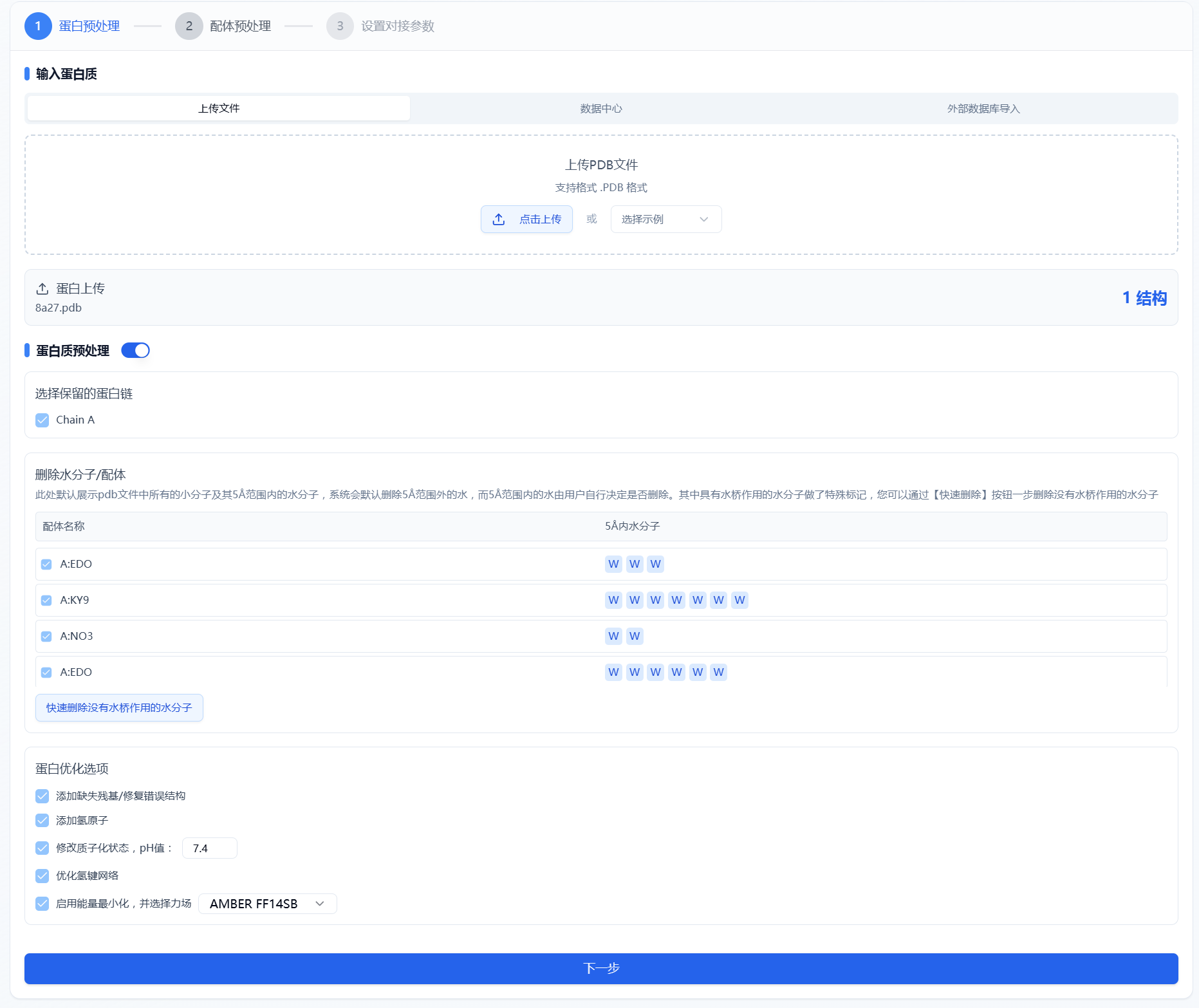
图 2. 蛋白预处理
- 输入蛋白质
平台为用户提供了三种上传蛋白文件的方式,分别为上传文件、数据中心和外部数据库导入。
上传文件。 复选框选中“上传文件”,通过点击上传按钮选择本地文件即可。上传的文件只支持.pdb 格式。
数据中心。复选框选中“数据中心”,通过点击下方从数据中心选择蛋白质,点击文件名称来选择数据中心的数据,点击完之后弹窗消失。
外部数据库导入。 复选框选中“外部数据库导入”,是在用户已知蛋白 PDB ID 的情况下,可直接在文本框中输入 4 位数的 PDB ID,即可下载并显示该蛋白结构。
注意!当前 CarsiDock 支持基于水分子的对接,暂不支持其它辅因子的对接。
- 蛋白质预处理
根据您上传的蛋白类型来确定是否进行蛋白预处理。
如果上传的蛋白已经做过蛋白预处理,您可以关闭蛋白质预处理后的开关,并直接点击下一步。
如果没有,建议您打开开关进行预处理相关的操作。
选择保留的蛋白链:此处默认显示上传的.pdb 文件中所有链的信息,默认所有链都被勾选。当某条链被取消勾选后,将不再参与最终的计算。
删除水分子/配体:此处默认展示 pdb 文件中所有的小分子及其 5Å 范围内的水分子。系统会默认删除 5Å 范围外的水,而 5Å 范围内的水由用户自行决定是否删除。其中具有水桥作用的水分子做了特殊标记,您可以通过【快速删除没有水桥作用的水分子】按钮一步删除没有水桥作用的水分子。删除某个小分子,需要取消该小分子前方的勾选。
- 蛋白优化选项
添加缺失残基/修复错误结构:可选项,默认为勾选状态;
添加氢原子:必选项;
修改质子化状态:可选项,默认为勾选状态,且 pH 值为 7.4;
优化氢键网络:可选项,默认为勾选状态;
启用能量最小化:可选项,默认为勾选状态,且选择的为 AMBER FF14SB 力场;
AMBER FF14SB.(推荐使用) FF14SB 是 AMBER 程序包中的一个蛋白质力场参数集,用于描述生物分子中的原子相互作用。它是 AMBER14 程序包中的一种特别适用于蛋白质体系的力场参数集,包括描述蛋白质中氨基酸侧链和蛋白质折叠中重要残基之间相互作用的额外参数。AMBER FF14SB 在描述蛋白质的构象和动力学性质方面具有较高的精度和可靠性。
AMBER FF15IPQ. FF15IPQ 是 AMBER 程序包中的一种改进的蛋白质力场参数集,相比于 AMBER FF14IPQ 具有更高的精度和可靠性。AMBER FF15IPQ 包括更多的偏极化效应和氢键参数,可以更准确地描述蛋白质的电子结构。
AMBER96. AMBER96 是 AMBER 程序包中的早期版本,经过多年的发展和优化,现在已有更新的版本,如 AMBER14 和 AMBER18 等。但是,AMBER96 仍然被广泛应用于生物分子模拟领域,特别是对于早期的研究和一些经典案例的模拟。
AMBER99SB. AMBER99SB 是 AMBER99 力场的一个改进版本,包括了描述蛋白质中氨基酸侧链和蛋白质折叠中重要残基之间相互作用的额外参数。AMBER99SB 在描述蛋白质的构象和动力学性质方面具有较高的精度和可靠性。
CHARMM36. CHARMM36 在描述蛋白质的构象和动力学性质方面具有较高的精度和可靠性。它在蛋白质和蛋白质-配体相互作用等领域的研究中被广泛应用,是生物分子模拟领域中常用的力场参数集之一。
(2) 配体预处理
平台提供了常规的小分子预处理方式,包括去除盐离子、保留最大的分子碎片、生成异构体(离子状态、互变异构体、立体异构体)和加氢、能量最小化。在我们平台上配体预处理的面板设置如图 3 所示。页面上默认选择的均为当前最优参数,用户也可根据自己的知识去选择适合的参数。此处参数与“配体预处理”模块一致。
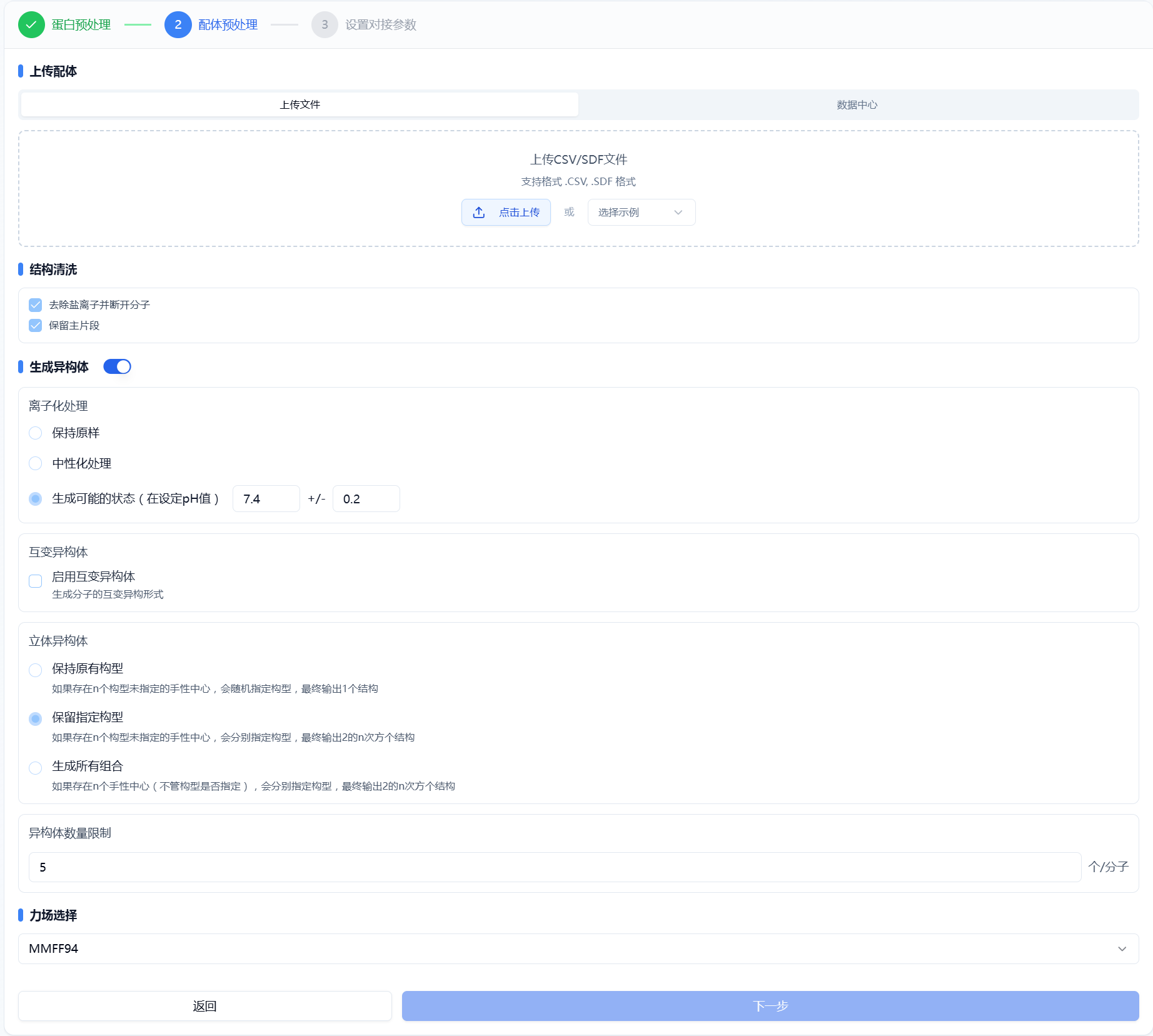
图 3. 配体预处理
- 上传配体
平台目前只支持以下方式上传配体。
上传文件。 复选框选中“上传文件”,通过点击下方按钮选择本地文件即可。文件格式支持.csv和.sdf。
数据中心。复选框选中“数据中心”,通过点击下方按钮页面出现弹窗,点击文件名称来选择数据中心的数据,点击完之后弹窗消失。
- 结构清洗
对上传结构进行处理。包括去除盐离子并断开分子、保留主片段。
- 生成异构体
当前平台默认对上传的分子进行枚举【开关状态为“开”】,以生成更多的异构体,包括离子化处理,互变异构体和立体异构体。当开关状态为“关”时,系统将不对分子做其他处理,保留分子的原始构型。
离子化处理。通过调整 pH 值范围,生成可能的离子化状态;
互变异构体。根据离子化状态,生成可能的互变异构体;
立体异构体。基于分子的手性特征,生成可能的立体异构体;
异构体数量限制。 平台默认最多输出 5 个异构体。用户可以根据自己需要调整。
- 力场选择
MMFF94. MMFF 是 Merck Molecular Force Field 的缩写,它是一个专业的小分子力场,是由 Hagler 开发的第二代分子力场,是目前最准确的力场之一。
UFF. UFF 是 Universal Force Field 的缩写,它是一个涵盖整个元素周期表的通用力场,该力场计算结构、结合能的计算结果精度一般,仅在找不到适合的力场时凑合用。
(3) 设置对接参数
在该步骤,用户需要先选择对接方法,然后再基于选择的对接方式去做相应的参数设置。
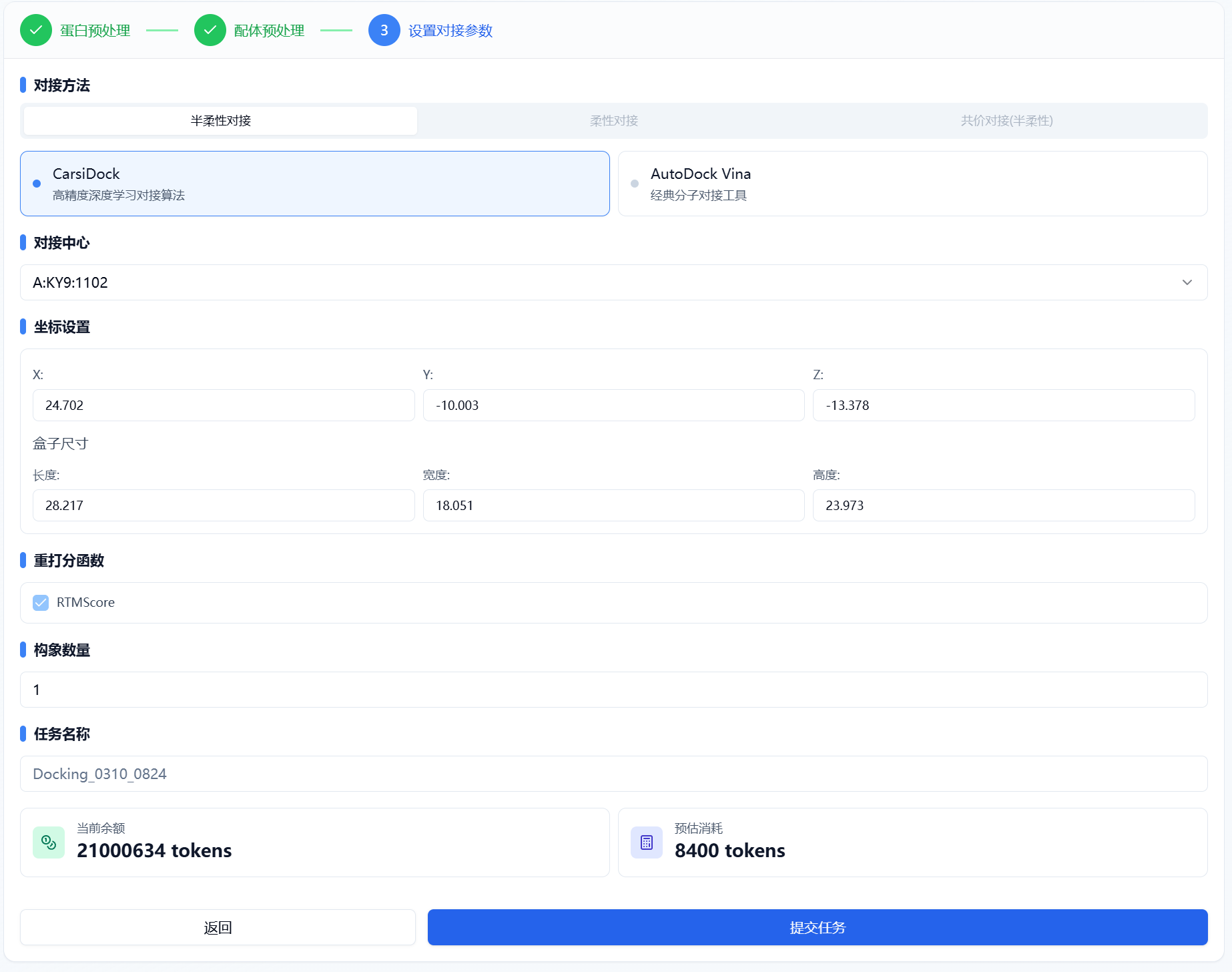
图 4. 设置对接参数
- 对接方法
目前平台支持三种不同类型的对接,包括半柔性对接、柔性对接、共价对接。
半柔性对接,受体构象保持固定,允许配体在结合口袋中进行构象采样与优化。平台提供两种方法:CarsiDock 是碳硅智慧自研的 AI 对接算法,速度快,精度高;而 AutoDock Vina 则是开源的软件,在此基础上我们做了一个优化,速度和精度比初始版本的要更好。
柔性对接,基于 Boltz-2 的柔性对接工具,在对接过程中同时考虑受体与配体的构象变化,更真实地模拟结合过程。相比半柔性对接,该方法通常具有更高精度,但计算开销也更大。
共价对接,用于模拟配体与受体之间形成共价键的结合模式,基于碳硅智慧自研的 CarsiDock-Cov 算法,支持 10 种常见共价反应类型。使用时需指定:共价残基和反应类型。
- 对接位点
CarsiDock 和 AutoDock Vina 均支持两种方式定义对接位点,分别为选择复合物中的配体和自定义对接位点。默认看到的坐标是系统随机分配,长宽高为系统默认值,这几个参数代表了结合口袋的位置和大小。
对接中心。 可以选择复合物中自带的配体作为对接中心。当复合物中有多个配体的时候,系统默认显示分子质量最大的配体,用户可以在下拉框中切换其他配体。系统会根据所选配体的大小计算出其几何中心(XYZ 坐标),并在配体最边缘原子上+5Å,计算盒子尺寸。
自定义对接位点。 对接中心也可选择自定义。在自定义模式下,用户可直接把鼠标放入 3D 显示区,当您单击某残基后,参数面板上才会显示出该残基相应的坐标信息,同时系统也给了一个默认的长宽高。但是,口袋具体的大小需要用户在了解蛋白口袋的情况下自行设置,口袋太小会导致计算结果不准确,口袋太大将会增加计算的时间。
- 重打分函数
采用碳硅智慧自研的重打分函数 RTMScore 对结果进行重打分,默认勾选。
- 构象数量
默认 1 个异构体输出 1 个对接构象,用户可通过输入框自行调整输出的数量,最多输出 100 个。
(4) 运行进度和结果查看
提交任务后,页面会自动跳入当前页面的“结果查看”子页面中,您可以在该页面查看当前模块的任务运行状态(进度条),也可在右上角的“通知”下拉框中查看所有模块正在运行的任务。当数据量较大时,系统会分批计算,因此只要有一批数据算完后(整个任务还在运行中),即可点击“查看结果”按钮进入结果页面,查看当前已完成计算的预测结果列表(未完成计算的分子暂不显示),并且可以在当前页面通过刷新来获取最新算完的数据。
图 5. 查看结果
3. 结果分析
结果页面由顶部的 Summary、左边的分子列表和右边的蛋白可视化区域组成。默认状态下,左边的结果详情区展示的是列表页面,可以切换至网格页面。其中,网格页面提供了一个简洁的查看分子结构的页面,而列表页面则提供了详细的计算结果,方便您分析当前的数据。 蛋白可视化区域是一个固定的内容,无论左边在什么子页面,蛋白可视化区域都将展示蛋白-配体的对接模式,用户可通过可视化区最下方的“<”、“>”快速浏览不同分子间的相互作用。
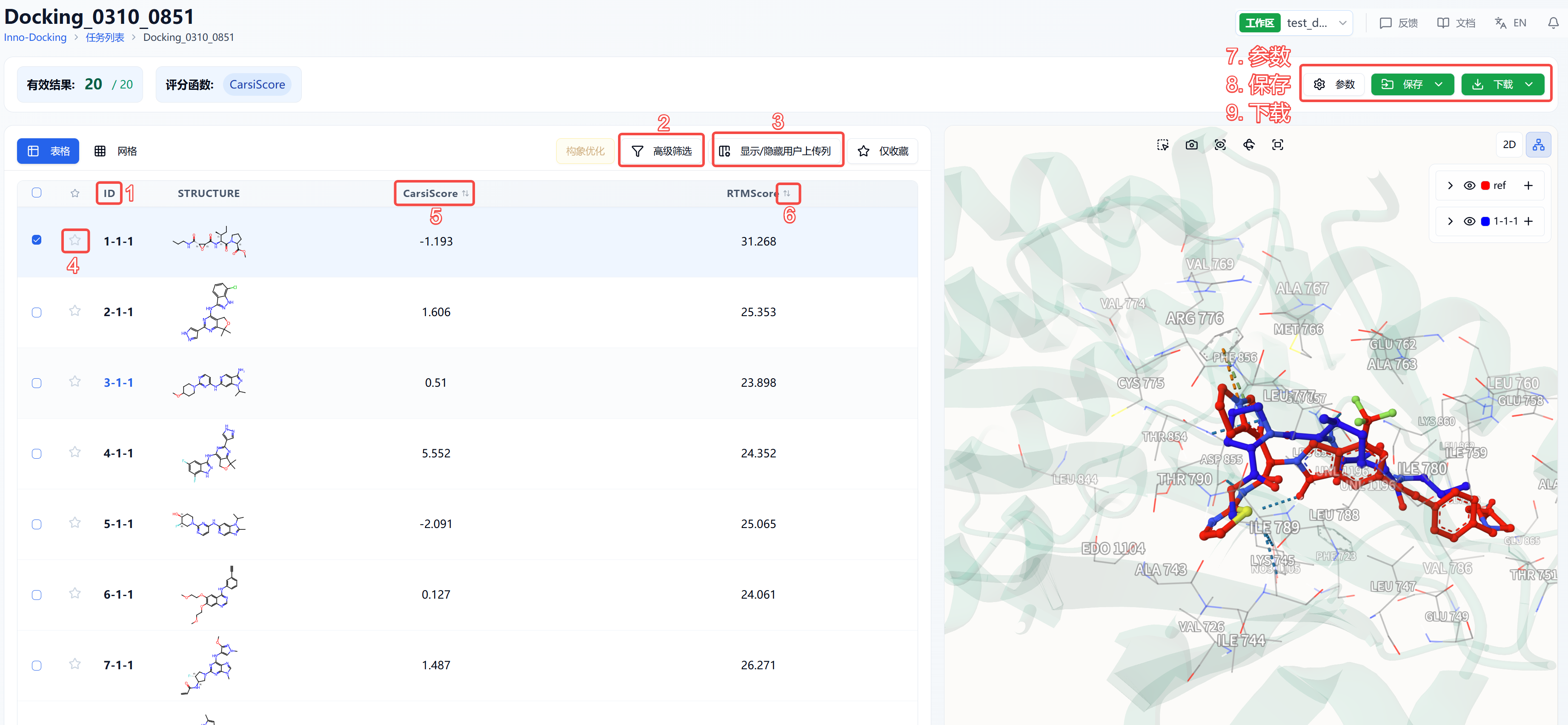
图 6. 结果页面功能分布
(1) 表格中 ID 的含义
ID 是根据原始文件的分子顺序分配的。如任务不生成异构体的情况下,ID 为 1-N 的连续值;如任务选择生成异构体,则 ID 将为 X-Y-Z 的组合,X 表示分子的顺序,Y 表示分子的异构体数量,Z 表示输出的构象数量,其中 Y 值越小,代表这个异构体存在的可能性越大。
(2) 高级筛选
高级筛选提供了范围筛选,可进一步筛选出某性质指定范围内的分子,以排除不符合预期结果的分子。进行高级筛选后,页面上只会显示符合过滤条件的分子。
(3) 显示/隐藏用户上传列
默认的结果列表不展示上传文件中的信息。根据显示/隐藏用户上传列中的选择,会更新结果列表的显示。在顶部还提供了两个快捷键“全选”和“全不选”,方便用户快速选择。
(4) 收藏
该功能主要用于帮助用户标记喜欢的分子。当您点击收藏某个分子后,该图标将会被点亮,意味着该分子被标记为收藏分子。点击表格右上角仅收藏之后,页面将只会显示被收藏的分子。点击收藏以后,可以再次点击该图标取消收藏。
(5) 属性解释
把鼠标移入每个性质的名称上,可查看该属性对应的解释。
(6) 排序
点击结果列表中的性质名字可重新排序,如 RTMScore,点击一次为升序,再点一次为降序,再点一次即恢复原始排序。
(7) 参数
显示对接任务的参数详情。
(8) 保存
点击“保存”,系统将弹出下拉框让您选择保存的文件,保存相应的数据至数据中心。保存的内容包括“预处理结果”和“对接后结果”。其中“预处理结果”指经过蛋白预处理和配体预处理但并没有对接的结构;“对接后结果”指经过对接程序输出的结果。“对接后结果”保存范围有三个选项:勾选的行,仅保存结果表格中首列方框勾选的分子;筛选后的行,仅保存经过高级筛选后得到的分子(页面左上角显示的有效结果可以看到有多少分子通过了高级筛选);所有行,保存输出的所有结果。
(9)下载
点击“下载”,系统将弹出下拉框让您选择下载的文件,下载相应的数据至本地。下载的内容包括“预处理结果”和“对接后结果”。其中“预处理结果”指经过蛋白预处理和配体预处理但并没有对接的结构;“对接后结果”指经过对接程序输出的结果。“对接后结果”下载范围有三个选项:勾选的行,仅下载结果表格中首列方框勾选的分子;筛选后的行,仅下载经过高级筛选后得到的分子(页面左上角显示的有效结果可以看到有多少分子通过了高级筛选);所有行,下载输出的所有结果。
4. 相关算法介绍
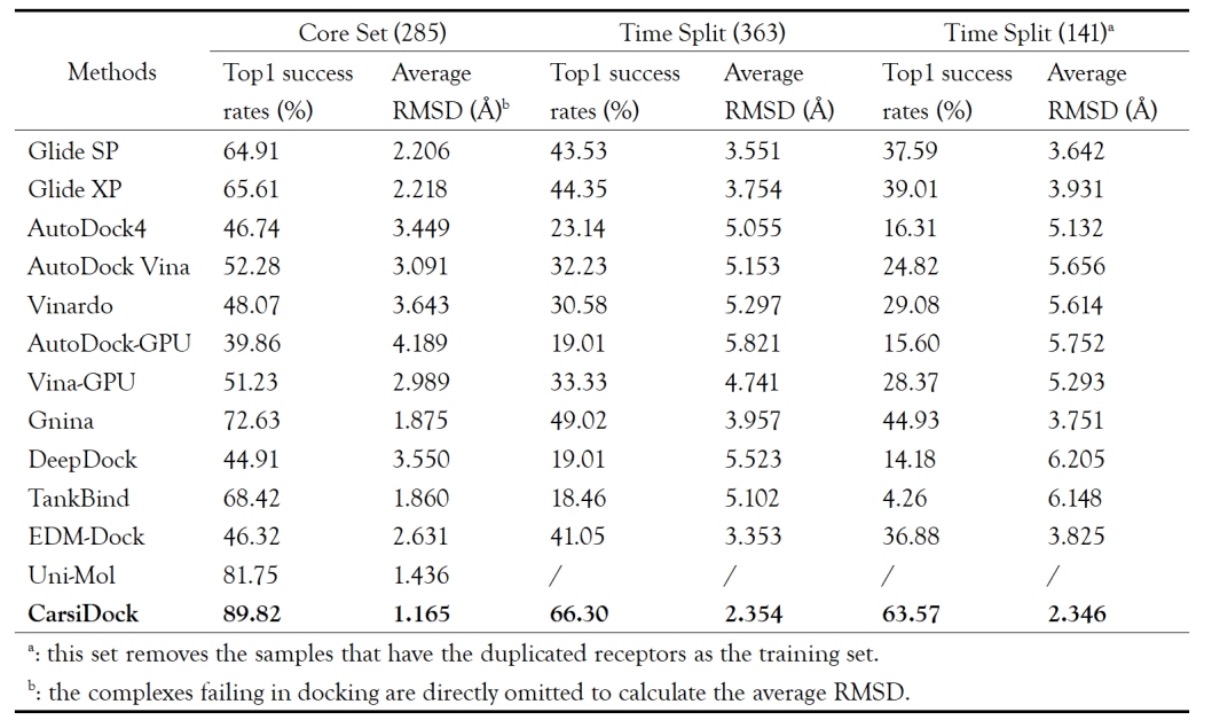
CarsiDock 是由碳硅智慧研发的半柔性 AI 对接方法,其首创了刚性对接指导的自蒸馏方法,并基于大规模物理模拟数据进行预训练。CarsiDock 该方法受启发于 BERT/ChatGPT 等预训练方法,对物理对接与 AI 对接方法进行了有效结合:首先使用 AI 方法预测蛋白与小分子的结合模式,再使用梯度下降快速得到对接构象。且使用物理对接方法生成大量的对接数据,这些物理模拟数据包括分子的结构、作用力模式、结合亲和力等重要信息,基于这些数据进行预训练,为 AI 对接提供了一个泛化能力强的底座模型,并利用晶体数据微调进一步提升 AI 预测的准确性。结果表明,CarsiDock 不仅可以保证结合姿态的拓扑可靠性,还可以成功地再现结晶结构中的关键相互作用,突出了其优越的适用性,具有优秀的对接能力和筛选能力。
CarsiDock-Cov是基于深度学习的自动化共价对接方法,该方法在原有非共价对接工具CarsiDock的基础上,通过整合共价键形成约束、几何优化、反应基团识别与配体共价修饰等专用模块,实现了对共价结合配体的高效、自动化对接。在多个公开数据集上的验证表明,CarsiDock-Cov在结合姿态预测和虚拟筛选性能方面优于多种现有共价对接工具,尤其在复杂交叉对接场景中表现出更好的泛化能力。
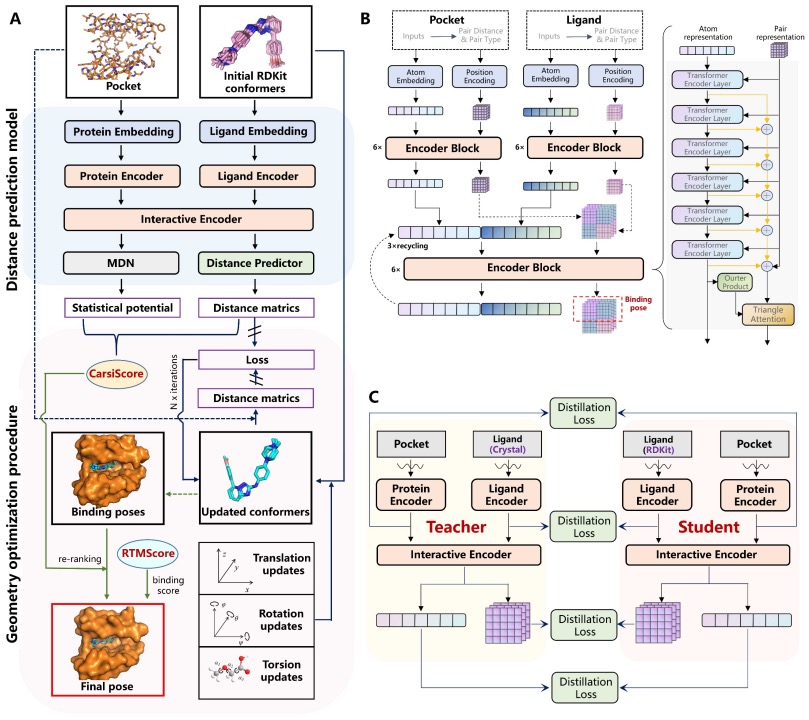
图 7. CarsiDock 模型框架
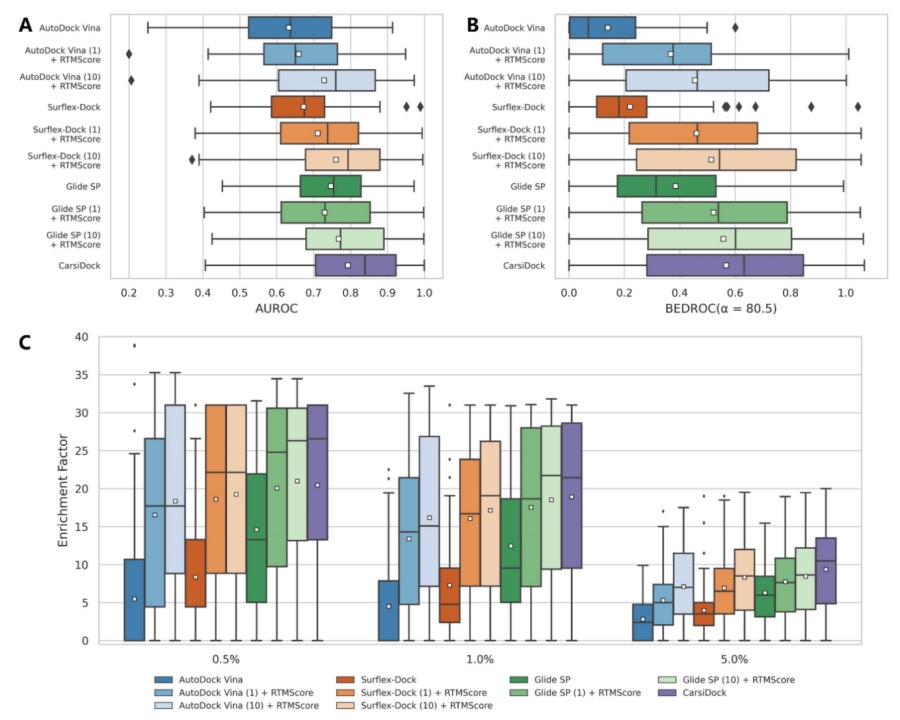
图 8. 多种方法在 DEKOIS2.0 数据集上的筛选性能
表 1. PDBBind CoreSet 及分时数据集的 Top1 成功率与 RMSD 平均值比较
5. 相关文献
【1】CarsiDock: a deep learning paradigm for accurate protein-ligand docking and screening based on large-scale pre-training, Chemical Science, 2024, 15, 1449-1471. 【2】CarsiDock-Cov: A deep learning-guided approach for automated covalent docking and screening, Acta Pharmaceutica Sinica B, 2025, 15, 5758-5771.