虚拟筛选
1. 虚拟筛选概述
虚拟筛选是指使用计算机技术在大量化合物库中搜索可能与某个药物靶点(通常是蛋白质受体或酶)结合的小分子的过程。这是药物设计中使用的一种重要技术,可以显著减少体外高通量筛选的时间和成本。传统药物筛选的最大缺陷在于基于对接做筛选速度慢,基于结构做筛选精度低。而本平台的虚拟筛选模块,采用速度最快的对接算法 KarmaDock 做第一级筛选,并采用精度最高的 CarsiDock 做第二级筛选,可以做到单台 8 卡服务器在 2 天内完成亿级别底库的筛选,在速度非常快的情况保证了筛选精度。其筛选速度是普通对接方法的数千倍,筛选精度也超过了领域内最常用的薛定谔的 Glide。
2. 使用说明
用户只需要四个步骤就可以完成计算:选择虚拟筛选数据库-上传蛋白质并确认口袋-自定义虚拟筛选流程-提交任务(必点)。
(1) 选择虚拟筛选数据库
虚拟筛选数据库是执行虚拟筛选的基础,数据库的质量直接影响到虚拟筛选的有效性。一方面,数据库需要包含足够多的分子,才能提供广阔的选择空间;另一方面,这些分子的信息需要准确无误,关于它们的化学性质、生物活性等信息都应是可靠的。从一个合理的数据库中寻找可能的药物候选分子,可大幅度缩短药物发现的时间,降低成本。
当前 DrugFlow 提供了多个不同类型的数据库,并且为了支持用户做大批量数据筛选,你可同时选择多个数据库进行计算。当前的数据库类型如下:
Enamine(4,076,929) :源自乌克兰的化合物品牌,化合物研发能力较强,有高性价比化合物和高价值化合物两类产品。
ChemDiv(1,648,184) :全球最大的化合物品牌之一,拥有 5000 多种化合物骨架结构和 100 多种化合物库,性价比高。
ChemBridge(1,634,292):源自美国的化合物品牌,拥有多样性库、大环库等多种热门化合物库。
TopScience Core Set(1,441,405) :为 1 万+活性小分子与 ChemDiv 的集合,具有结构组成多样、化合物多样性好、类药性佳以及高性价比等优点。使用该数据库可以在合适的筛选范围内发现高质量、高性价比的化合物,减少初期研究的成本,同时满足药物功能重定位、虚拟筛选最核心的筛选需求。
TopScience Refine Set(7,172,703) :包含 8 个品牌,其中包括常用的Enamine、ChemBridge、Maybridge 等数据库。该数据库在核心数据库的基础上增加了大量的化合物结构,为科学家们提供更多筛选可能性。适合需要大量结构数据,且经费充足的用户使用。为精选数据库,极大增加了可选化学空间。
TopScience Extend Set(5,102,060):包含 14 个品牌,包括大家熟知的Alinda Chemical、Asinex、Innovapharm等数据库品牌。对于算力足够的虚拟筛选来说,可获得实体化合物的结构数量总是难以满足需求。该数据库在前述数据的基础上扩展收集了更多数据,使数据库包含更丰富多样的结构类型。拥有扩展的类药化合物结构,为筛选提供更多可能性。
Drug Repurposing Compound Library(4,317):药物功能重定位化合物库收集了 4317 种已上市和进入临床期的药物,这些化合物经过大量的临床前研究实验,具有活性高、毒性低和机制明确的特点,适用于药物筛选的同时,也是细胞诱导分化的有力工具。
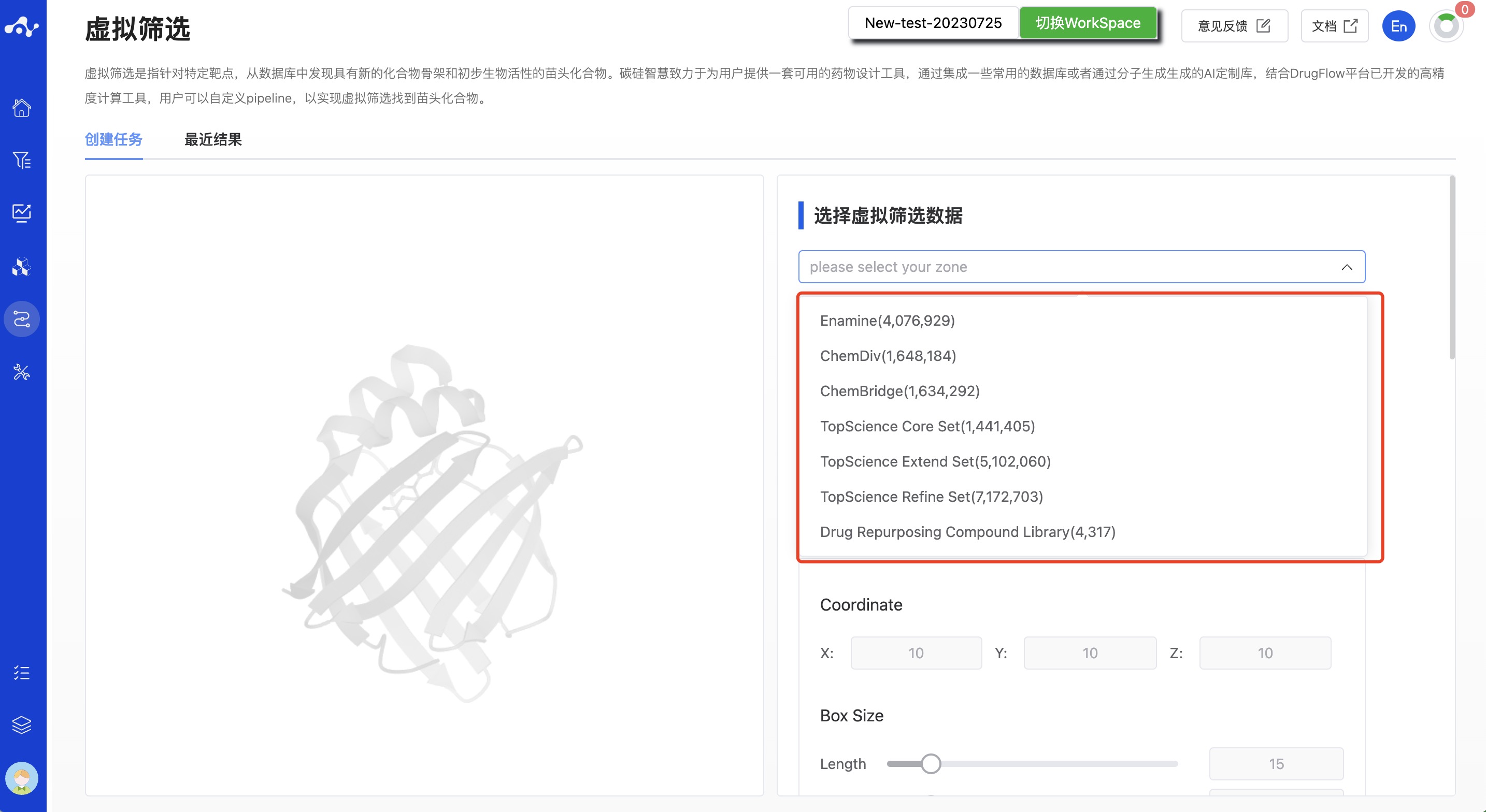
图 1. 平台支持的数据库类型
注意:当前针对试用用户,仅开放药物库,而付费用户则可以支持高达 2000 万的底库(可多选)。
(2) 上传蛋白质并确认口袋
除了选择虚筛数据库,还有一个关键的对象——研究靶点。目前我们的虚拟筛选以对接为主要功能,因此进行虚拟筛选之前,需要上传研究靶点的蛋白文件并指定对接口袋。上传蛋白文件的方式有两个,分别为上传文件或从数据中心选择。
- 上传文件
复选框选中“上传文件”,通过点击下方按钮选择本地文件即可。选择完文件以后,该文件名称将显示在按钮上,右边将显示文件内容。 关于上传的文件只支持.pdb 格式。
- 数据中心
复选框选中“数据中心”,通过点击下方按钮页面出现弹窗,点击文件名称来选择数据中心的数据,点击完之后弹窗消失,即可提交任务。
(3) 自定义虚拟筛选流程
在最终的虚拟筛选模块上,我们将为用户提供一套自定义 pipeline 的流程,用户可以应用虚拟筛选模块,应用 DrugFlow 已上线的功能来自定义 pipeline,并设置合理的过滤条件,即可一键发起虚拟筛选任务,用户只需要等待这个 pipeline 算完,就可以得到结果,然后再去基于得到的结果进一步的分析,就能获得理想的活性分子。
针对当前的版本,我们目前仅支持既定的流程,即 Inno-ADMET > KarmaDock > CarsiDock.
步骤 1. Inno-ADMET
Inno-ADMET 步骤主要基于成药性性质对分子做过滤。默认参数下,平台会使用 MW、TPSA、Log P 和 Log S 四个性质进行过滤。用户可以随时调整过滤条件,或者通过下拉的方式更换,也可以删除和新增过滤属性。
根据 Inno-ADMET 的过滤条件得到的分子,将进入 KarmDock 中进行计算。
注意!请注意您的过滤条件,避免所有分子都被过滤的情况。
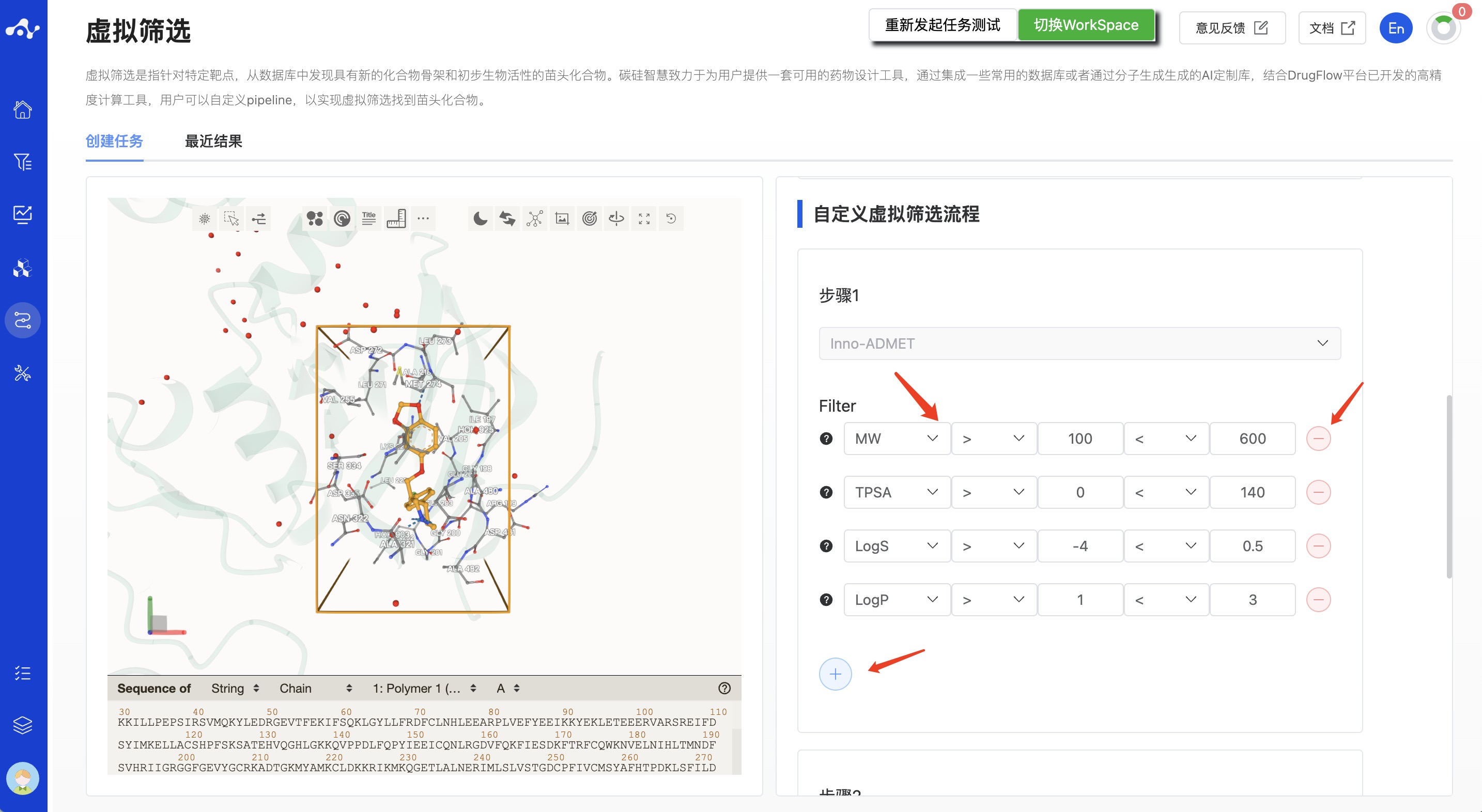
图 2. 设置 ADMET 过滤参数
步骤 2. KarmaDock
KarmaDock 是碳硅智慧自研的 AI 对接算法,它通过打分与对接的多阶段学习,在轻量化的模型上有效结合了对接和打分方法。在保证了对接与虚筛性能的同时,将单个小分子对接+打分的耗时压缩至 17ms,远低于其他模型,且其精度也处于领先水平(详情可见“相关算法介绍”)。因此我们采用速度最快的 KarmaDock 作为第二步过滤,KarmaDock 将输出 KarmaScore 值,该值越大,代表其结合亲和力越好。基于此,系统应用 KarmaScore 进行降序排序,将根据你设定的数值来保留排名前多少的数据,且该部分数据将进入下一步 CarsiDock 的计算。
步骤 3. CarsiDock
CarsiDock 也是碳硅智慧自研的 AI 对接算法,它是一个高精度对接算法,被安排在了虚筛的第三步。它当前的对接速度,优化到了 2s/分子。由于 CarsiDock 对构象的比较敏感,因此在此步骤中,针对 KarmaDock 保留的分子,我们建议对分子生成立体异构体。CarsiDock 计算可以得到 CarsiScore 和 RTMScore。其中,CarsiScore 是一个构象打分值,该值越小越好,而 RTMScore 是一个结合亲和力的值,该值越大越好。因此此处以 RTMScore 作为过滤对象,你可以选择取 RTMScore 超过某一个阈值的分子,也可以基于 RTMScore 降序,取前 XXX 的分子或者前 XX%的分子。
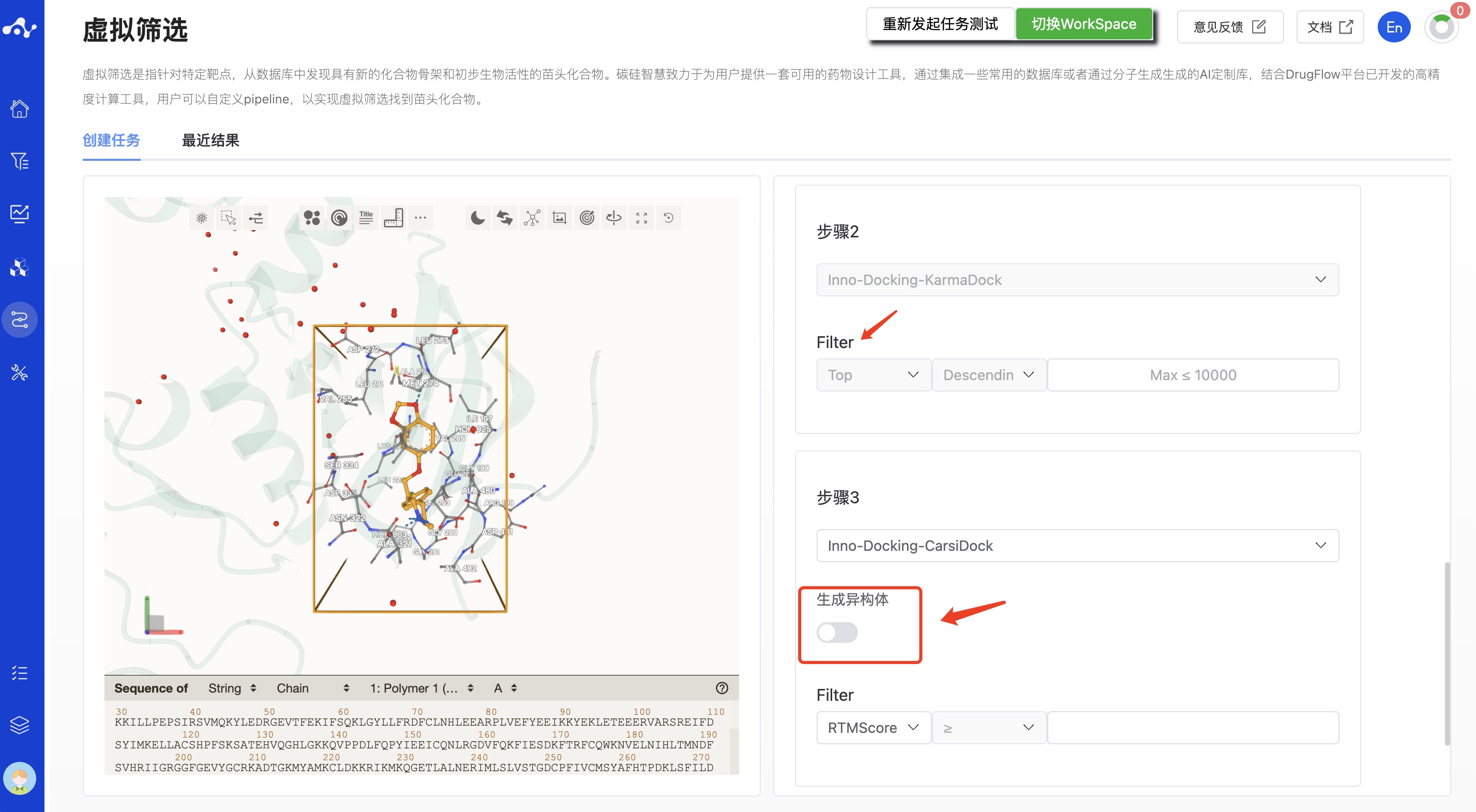
图 3. 设置 KarmaDock 和 CarsiDock 的过滤参数
(4) 运行进度和结果查看
提交任务后,页面会自动跳入当前页面的“最近结果”子页面中,你可以在该页面查看当前模块的任务运行状态(进度条),也可在右上角的“运行中“下拉框中查看所有模块正在运行的任务。当数据量较大时,系统会分批计算,因此只要有一批数据算完后(整个任务还在运行中),即可点击“结果详情”按钮进入结果页面,查看当前已完成计算的预测结果列表(未完成计算的分子暂不显示),并且可以在当前页面通过刷新来获取最新算完的数据。
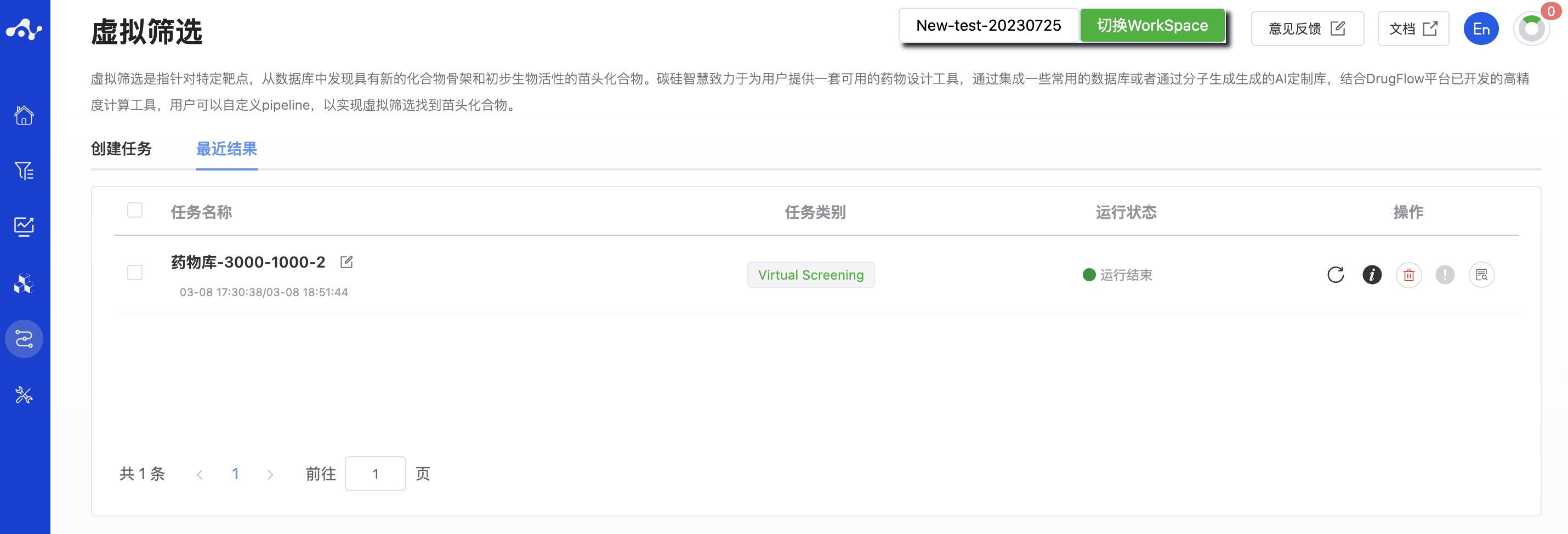
图 4. 查看结果
3. 结果分析
结果页面由顶部的 Summary、左边的分子列表和右边的蛋白可视化区域组成。默认状态下,左边的结果详情区展示的是网格页面,可以切换至列表和聚类子页面。其中,网格子页面提供了一个简洁的查看分子结构的页面,而列表页面则提供了详细的计算结果,方便你分析当前的数据,而聚类子页面则将得到的分子按照 Murcko 骨架对分子进行分类展示。 蛋白可视化区域是一个固定的内容,无论左边在什么子页面,蛋白可视化区域都将展示蛋白-配体的对接模式,用户可通过右下角的“上一个”、“下一个”快速浏览分子间的相互作用。
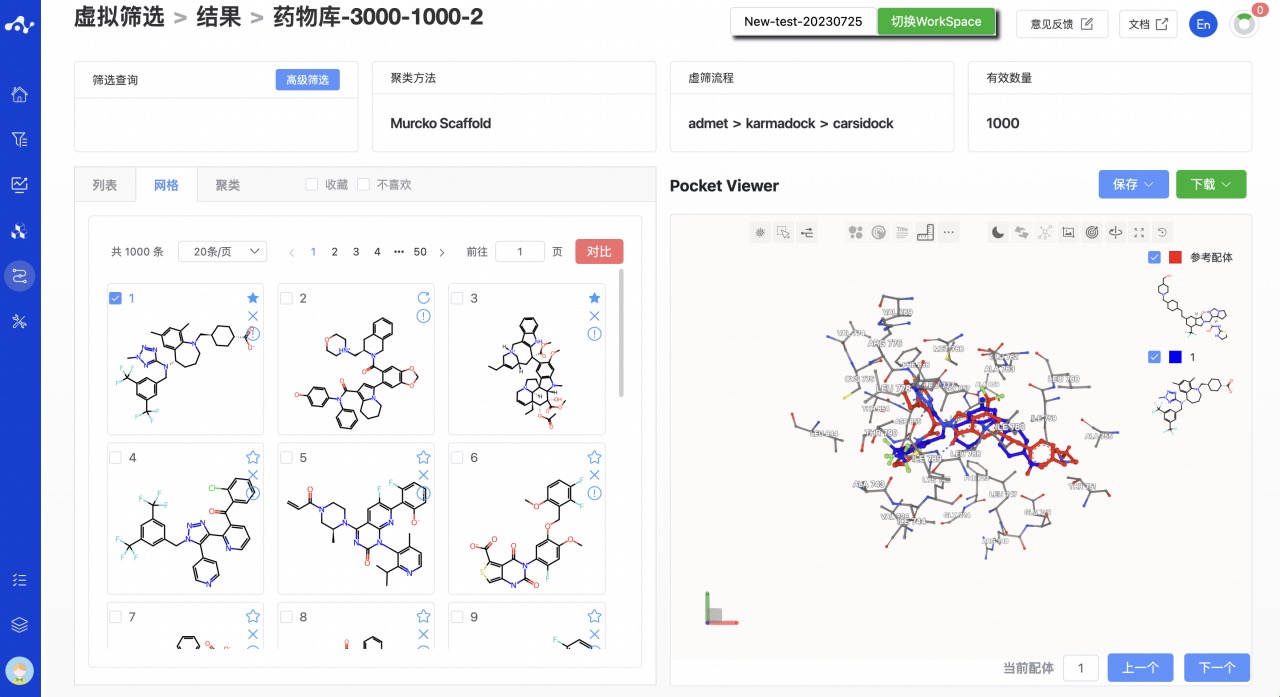
图 5. 虚拟筛选结果页面-默认网格页面
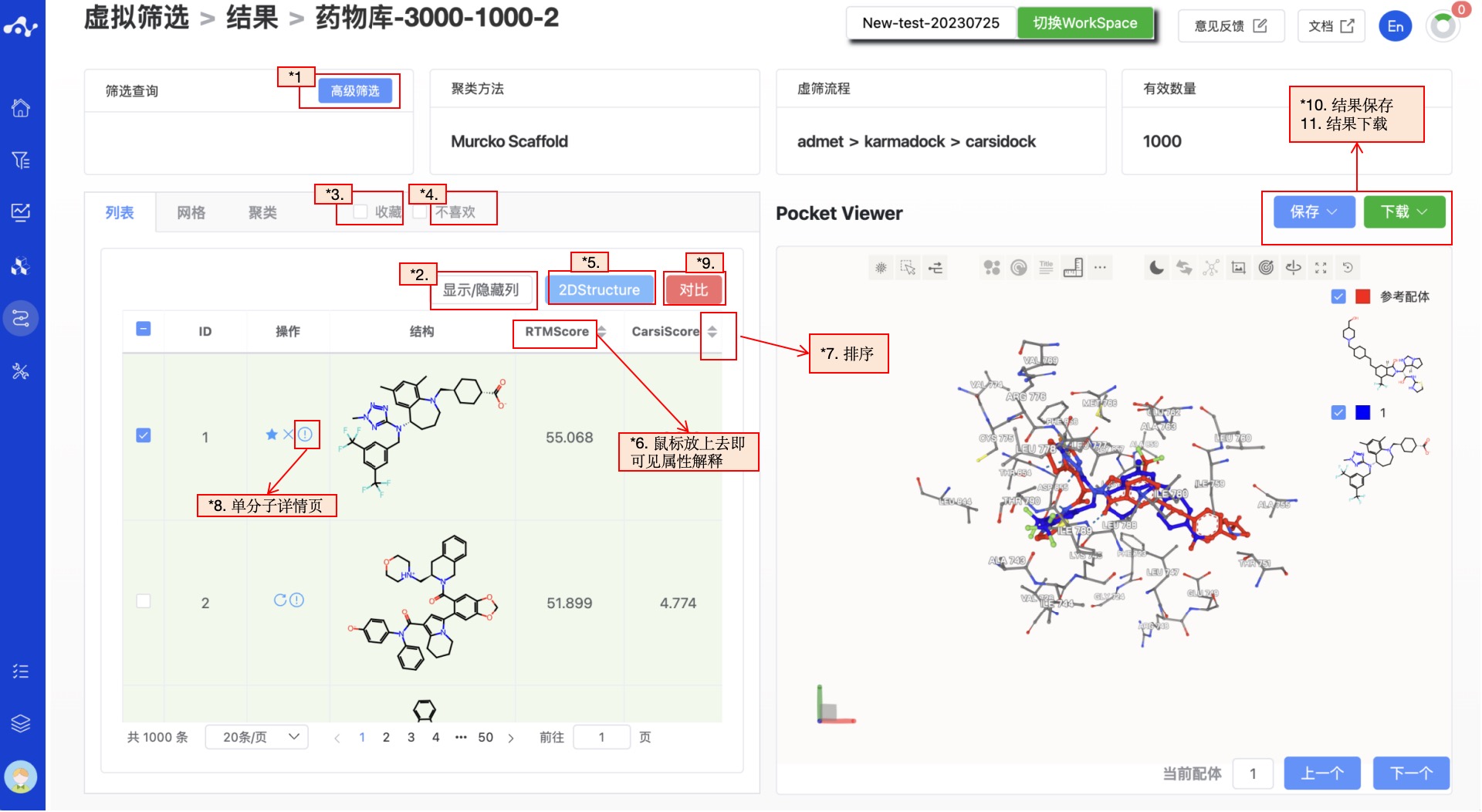
图 6. 结果页面功能布局-列表页面
(1) 高级筛选
高级筛选提供了范围筛选,可进一步筛选出某性质指定范围内的分子,以排除不符合预期结果的分子。进行高级筛选后,页面上只会显示符合过滤条件的分子。
(2) 显示/隐藏上传列
默认的结果列表不展示上传文件中的信息,此时左边控制栏中为不选中的状态。当你不想显示该性质时,取消该性质的选中即可,左边的结果列表将根据控制栏中的选择进行实时显示。在顶部还提供了两个快捷键“全选”和“不选”,方便用户快速选择。
(3) 收藏
该功能主要用于帮助用户标记喜欢的分子。当你点击收藏某个分子后,该图标将会被点亮,意味着该分子被标记为收藏分子。点击收藏的勾选框之后,页面将只会显示被收藏的分子。点击收藏以后,可以再次点击该图表取消收藏。
(4) 不喜欢
该功能主要用于帮助用户标记不喜欢的分子。当点击不喜欢某个分子后,收藏的图标将消失,且不喜欢的图标会改成恢复图标,意味着用户可以随时修改该分子的标签。点击不喜欢的勾选框之后,页面将只会显示被标记为不喜欢的分子。
(5) 2D Structure
默认该按钮被选中,列表中正常显示配体结构,点击一次后可取消选中,则列表中该分子结构被隐藏。
(6) 属性解释
把鼠标移入每个性质的名称上,可查看该属性对应的解释。
(7) 排序
点击结果列表中的性质名字可重新排序,如 CarsiScore,点击一次为升序,再点一次为降序,再点一次即恢复原始排序。
(8) 单个分子详情页
点击“分子详情”按钮,页面将跳转至单分子详情页,该页面全面展示了分子的各种信息让用户快速了解该分子的详细信息,包括此次计算的内容,以及该分子的 Inno-ADMET 属性和对接构象。
- 点击 Inno-ADMET,可以看到当前分子的物化性质、药化性质、类药性、吸收、分布、代谢、排泄、和毒性等性质。将鼠标放在各项性质右侧,可以查看释义及推荐范围;
- 点击对接构象,可以看到当前分子与蛋白口袋结合时的的复合物构象。另外,也提供了参考分子的结合构象便于比较。

图 7. 虚拟筛选结果页面-单分子详情页
(9) 对比
每个分子的序号前面都有一个复选框,当你同时勾选多个分子时,即可点击【对比】按钮,页面即跳转到对比页面,方便进行多个分子的全面比较。该页面与单分子详情页比较类似,包含了该分子的 Inno-ADMET 属性、ChemFH 属性、Inno-SA 属性和对接构象。

图 7. 虚拟筛选结果页面-多分子对比页
(10) 保存
点击“保存”,系统将弹出下拉框让你选择保存的文件格式(目前仅支持.csv/.sdf)。确定好保存的文件样式后,保存相应的数据为 sdf 或 csv 文件至数据中心。保存的内容是页面展示的有效数量的分子,而这些分子通常是根据你的显示隐藏列的条件、高级筛选的条件、收藏或者是不喜欢来获取的。
(11)下载
点击“下载”,系统将弹出下拉框让你选择下载的文件格式(目前仅支持.csv/.sdf)。确定好下载的文件样式后,系统将下载相应的数据为 sdf 或 csv 文件至本地设备上。下载的内容与保存的方式一致,也是下载的页面展示的有效数量的分子,而这些分子通常是根据你的显示隐藏列的条件、高级筛选的条件、收藏或者是不喜欢来获取的。
4. 相关算法介绍
KarmaDock 是碳硅智慧自研的 AI 对接算法,它通过打分与对接的多阶段学习,在轻量化的模型上有效结合了对接和打分方法。在保证了对接与虚筛性能的同时,将单个小分子对接+打分的耗时压缩至 17ms。
Karmadock 应用编码器(Graph Transformer(GT)和 Geometric Vector Perceptrons(GVP),以及用于对接的 EGNN 模块和用于打分的混合密度网络 MDN 模块搭建的模型框架。建模原理:首先将蛋白和配体分别表征为三维残基图和二维分子图,使用 GVP/GT 编码器编码后计算 MDN 模块中混合高斯分布的 pi, mu, sigma,之后将图特征送入 EGNN 模块进行对接,得到对接构象,并将其送入 MDN 模块进行打分,得到 Score。其建模框架详见图 7.
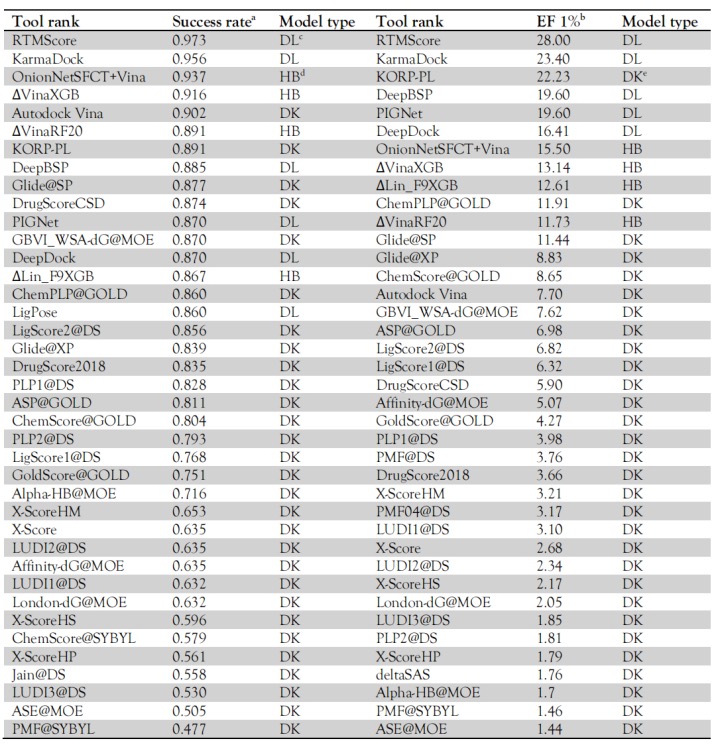
KarmaDock 是同时包含对接和打分的端到端 AI 模型。在单 GPU 上实现毫秒级响应,且在精度上超越大多数对接/打分工具。可实现超大规模分子库的高通量筛选,加速药物研发。KarmaDock 对接一个分子的平均耗时为 0.017s/complex,远低于其他模型。KarmaDock 在 Coreset 数据集上的对接成功率(RMSD<2 Å)为 0.956,超越绝大多数模型。并且 KarmaDock 在 Casf2016 数据集上的富集因子(EF1%)为 23.4,处于领先水平。详见表 1.
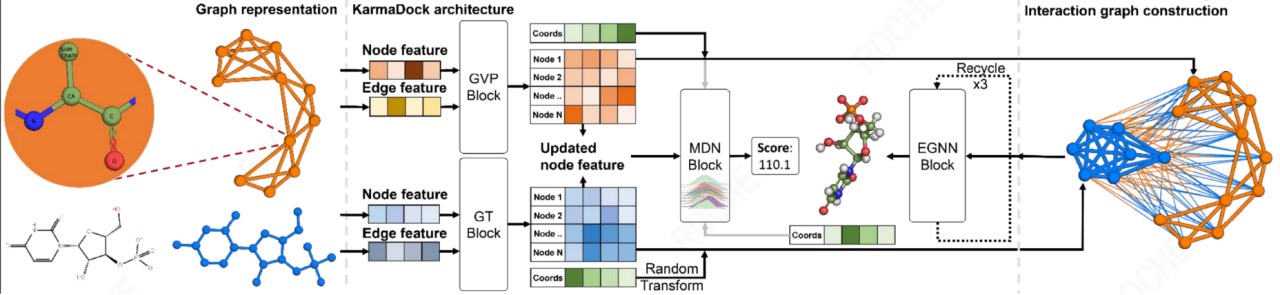
图 9. KarmaDock 建模框架
表 1. 各个模型在 CASF 2016 上的对接能力和筛选能力
注意:CarsiDock 和 RTMScore 的介绍,分别见 Inno-Docking 和 Inno-Rescoring.
5. 相关文献
[1] Efficient and accurate large library ligand docking with KarmaDock. Zhang X. Zhang O. Shen C., et al. Nat Comput Sci. 2023 Sep;3(9):789-804. doi: 10.1038/s43588-023-00511-5.