Virtual Screening
1. Overview of Virtual Screening
Virtual screening refers to the process of using computer technology to search for small molecules that may bind to a drug target (usually a protein receptor or enzyme) in large compound libraries. This is an important technique used in drug design that can significantly reduce the time and cost of in vitro high-throughput screening. The biggest drawback of traditional drug screening is that it is slow when screening is based on docking and accuracy is low when screening is based on structure. The virtual screening module of this platform uses KarmaDock, the fastest docking algorithm for first-level screening, and CarsiDock, the most accurate one for second-level screening, capable of completing billion-level screening in two days with a single 8-card server, ensuring screening accuracy in very fast scenarios. Its screening speed is thousands of times that of common docking methods, and its screening accuracy also exceeds that of Schrödinger's Glide, the most commonly used in the field.
2. Instructions for Use
Users only need four steps to complete the calculation: choose a virtual screening database - upload the protein and confirm the pocket - customize the virtual screening process - submit the task.
(1) Choose a Virtual Screening Database
The virtual screening database is the basis for executing virtual screening, and the quality of the database directly affects the effectiveness of virtual screening. On the one hand, the database needs to contain enough molecules to provide a wide range of choices; on the other hand, the information about these molecules needs to be accurate, and the information about their chemical properties, biological activity, etc., should be reliable. Finding possible drug candidate molecules from a reasonable database can greatly shorten the time for drug discovery and reduce costs.
Currently, DrugFlow provides several different types of databases, and to support users in conducting large-volume data screening, you can choose multiple databases for calculation simultaneously. The current database types are as follows: Enamine(4,076,929): Sourced from a Ukrainian compound brand, with strong compound research and development capabilities, it has two types of products: cost-effective compounds and high-value compounds.
ChemDiv(1,648,184): One of the world's largest compound brands, with more than 5,000 types of compound scaffold structures and over 100 compound libraries, offering high cost performance.
ChemBridge(1,634,292): Originating from a US compound brand, it offers various popular compound libraries such as diversity libraries, macrocycle libraries, etc. TopScience Core Set(1,441,405): A collection of 10,000+ active small molecules and ChemDiv, with diverse structure composition, good compound diversity, excellent drug-likeness, and high cost performance. Using this database can discover high-quality, cost-effective compounds within a suitable screening range, reduce the cost of early research, and meet the most core screening requirements of drug function repositioning and virtual screening.
TopScience Refine Set(7,172,703): It includes 8 brands, including commonly used databases such as Enamine, ChemBridge, Maybridge, etc. This database adds a large number of compound structures based on the core database, providing scientists with more screening possibilities. Suitable for users who need a large amount of structural data and have sufficient funding. It is a carefully selected database, greatly increasing the optional chemical space.
TopScience Extend Set(5,102,060): It includes 14 brands, including well-known database brands like Alinda Chemical, Asinex, Innovapharm, etc. For virtual screening with sufficient computing power, the number of physical compound structures that can be obtained is always difficult to satisfy. This database extends more data collection based on the aforementioned data, making the database contain more diverse structure types. It has extended drug-like compound structures, providing more possibilities for screening.
Drug Repurposing Compound Library(4,317): The Drug Repurposing Compound Library collects 4,317 approved and clinically entered drugs. These compounds have undergone extensive pre-clinical research and are characterized by high activity, low toxicity, and clear mechanisms. They are suitable for drug screening and are also powerful tools for cell-induced differentiation.
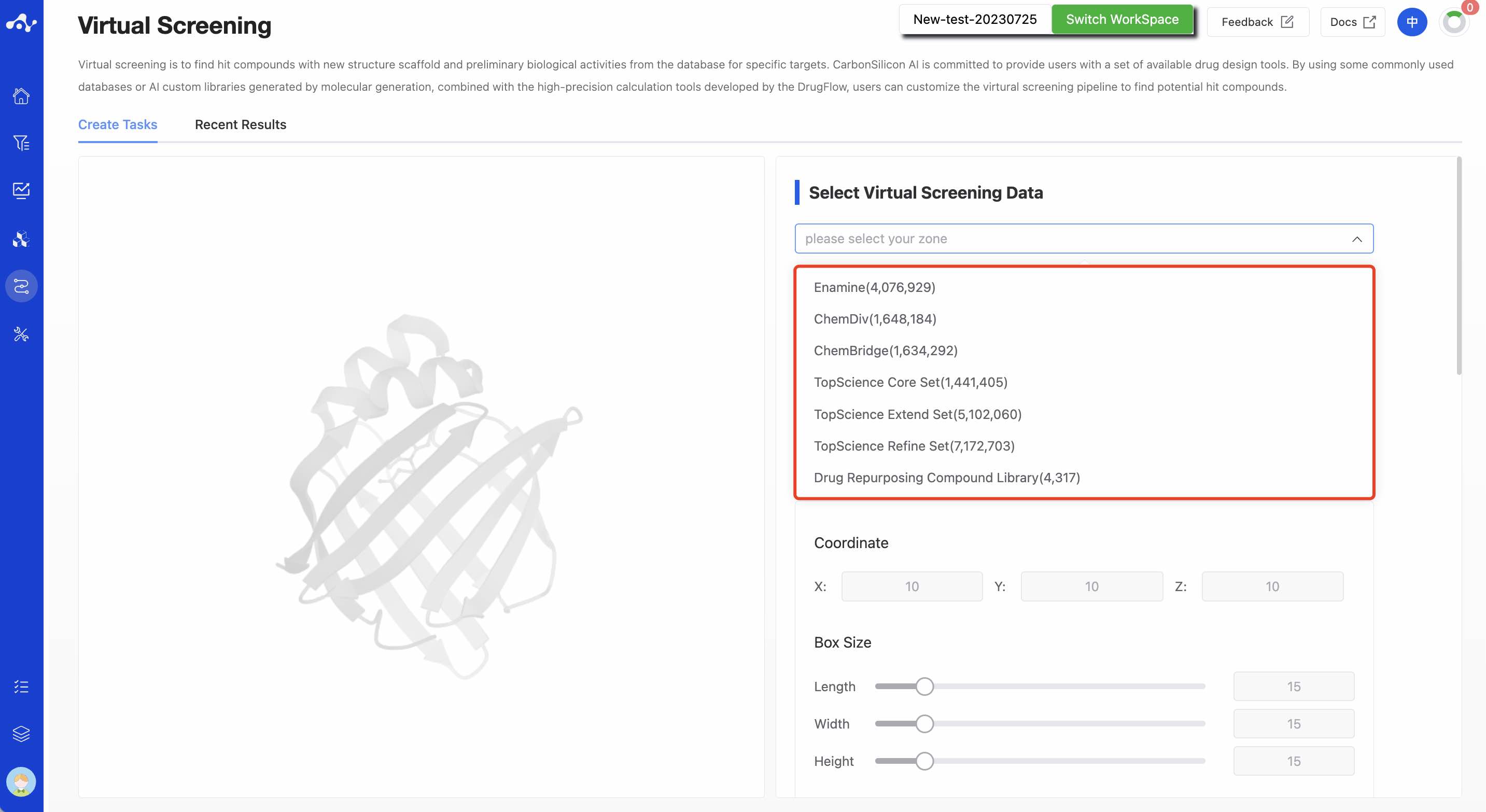
Figure 1. Types of databases supported by the platform Note: Currently for trial users, only the drug library is available, while for paid users, they can support up to 20 million base libraries (multiple selection allowed).
(2) Upload protein and set pocket (Optional field)
In addition to choosing a virtual screening database, there is another key object - the research target. Currently, our virtual screening mainly relies on docking, so before conducting virtual screening, you need to upload the protein files of the research target and designate the docking pocket. There are two ways to upload protein files, either by uploading files or by selecting from the data center.
- Protein Upload
The platform provides two methods of protein upload: file upload and data center. After the protein is uploaded, it will automatically be visualized on the left.
Upload file: Check the "Upload file" checkbox, and select the local file by clicking the button below. The currently supported file format is .pdb. After the file is selected, its name will be displayed on the button;
Data center: Select the "Data center" checkbox, and a popup will appear when you click the button below. You can select the data center's data by clicking on the file name, and the popup will disappear after clicking.
- Pocket Setting
Specifying the binding pocket of the molecule, there are two ways to do this: Based on the PDB file you uploaded, the system will automatically select the molecule with the largest molecular weight as the pocket, and determine the geometric center coordinates and Box Size based on this ligand molecule; you can also customize the pocket by manually adjusting the coordinates and Box Size of the pocket.
(3) Custom Virtual Screening Pipeline
In the virtual screening module, we will provide users with a custom pipeline process. Users can use the virtual screening module, use the functions that DrugFlow has already launched to customize the pipeline, and set reasonable filter conditions. Then, with one click, they can initiate a virtual screening task. Users just have to wait for this pipeline to finish its calculations, and then they can get the results. Further analysis can be made based on these results to get the desired active molecules.
For the current version, we only support a predetermined process, namely, Inno-ADMET > KarmaDock > CarsiDock.
Step 1. Inno-ADMET
The Inno-ADMET step is mainly based on drug-likeness properties to filter molecules. By default, the platform will use MW(Molecular Weight), TPSA (Total Polar Surface Area), Log P (Partition Coefficient) and Log S (Solubility) four properties for filtering. Users can adjust the filter conditions at any time, or change them using a drop-down method, and they can also delete and add new filter properties.
The molecules obtained from the Inno-ADMET filter conditions will be calculated in KarmaDock.
Note! Please pay attention to your filter conditions to avoid all molecules being filtered out.
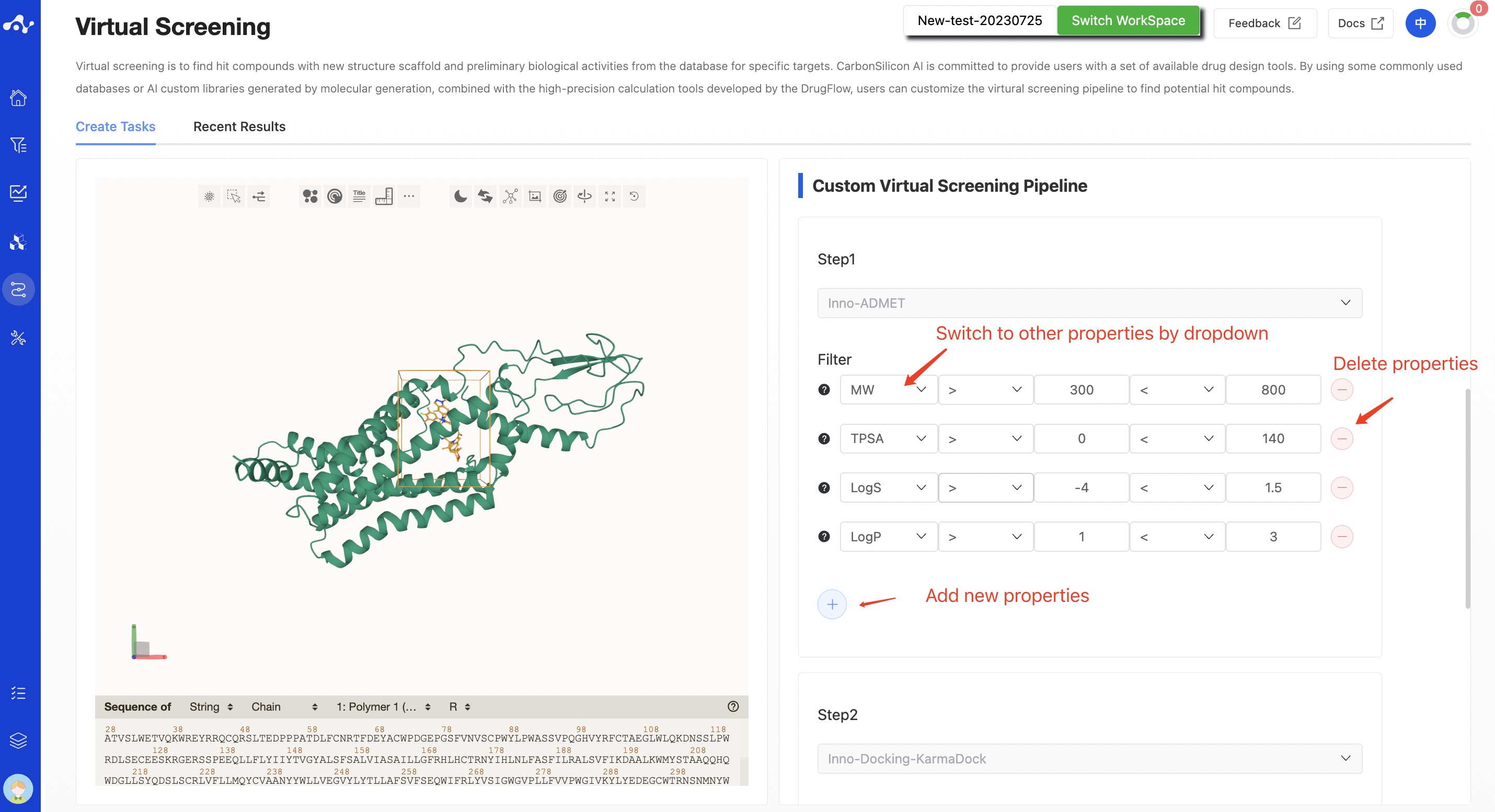
Figure 2. Setting ADMET Filtering Parameters
Step 2. KarmaDock
KarmaDock is an AI docking algorithm developed by Carbon Silicon Intelligence. It effectively combines docking and scoring methods on a lightweight model through multi-stage learning of scoring and docking. While ensuring docking and virtual screening performance, it compresses the time consumption of single small molecule docking+scoring to 17ms, which is far lower than other models, and its accuracy is also leading (details can be seen in "Related Algorithm Introduction"). Therefore, we use the fastest KarmaDock as the second step of filtering. KarmaDock will output a KarmaScore value. The larger the value, the better the binding affinity. Based on this, the system uses KarmaScore for descending order sorting, and it will retain the top data according to the value you set. This part of the data will be calculated in the next step of CarsiDock.
Step 3. CarsiDock
CarsiDock is also an AI docking algorithm developed by Carbon Silicon Intelligence. It is a high-precision docking algorithm and is arranged as the third step of virtual screening. Its current docking speed has been optimized to 2s/molecule. Since CarsiDock is sensitive to conformation, therefore, in this step, for the molecules retained by KarmaDock, we suggest generating stereoisomers for the molecules. CarsiDock calculation can obtain CarsiScore and RTMScore. Among them, CarsiScore is a conformation scoring value, the smaller the better, and RTMScore is a binding affinity value, the larger the better. Therefore, RTMScore is used as the filtering object here, you can choose to take molecules with RTMScore above a certain threshold, or you can sort in descending order based on RTMScore, taking the top XXX molecules or the top XX% molecules.
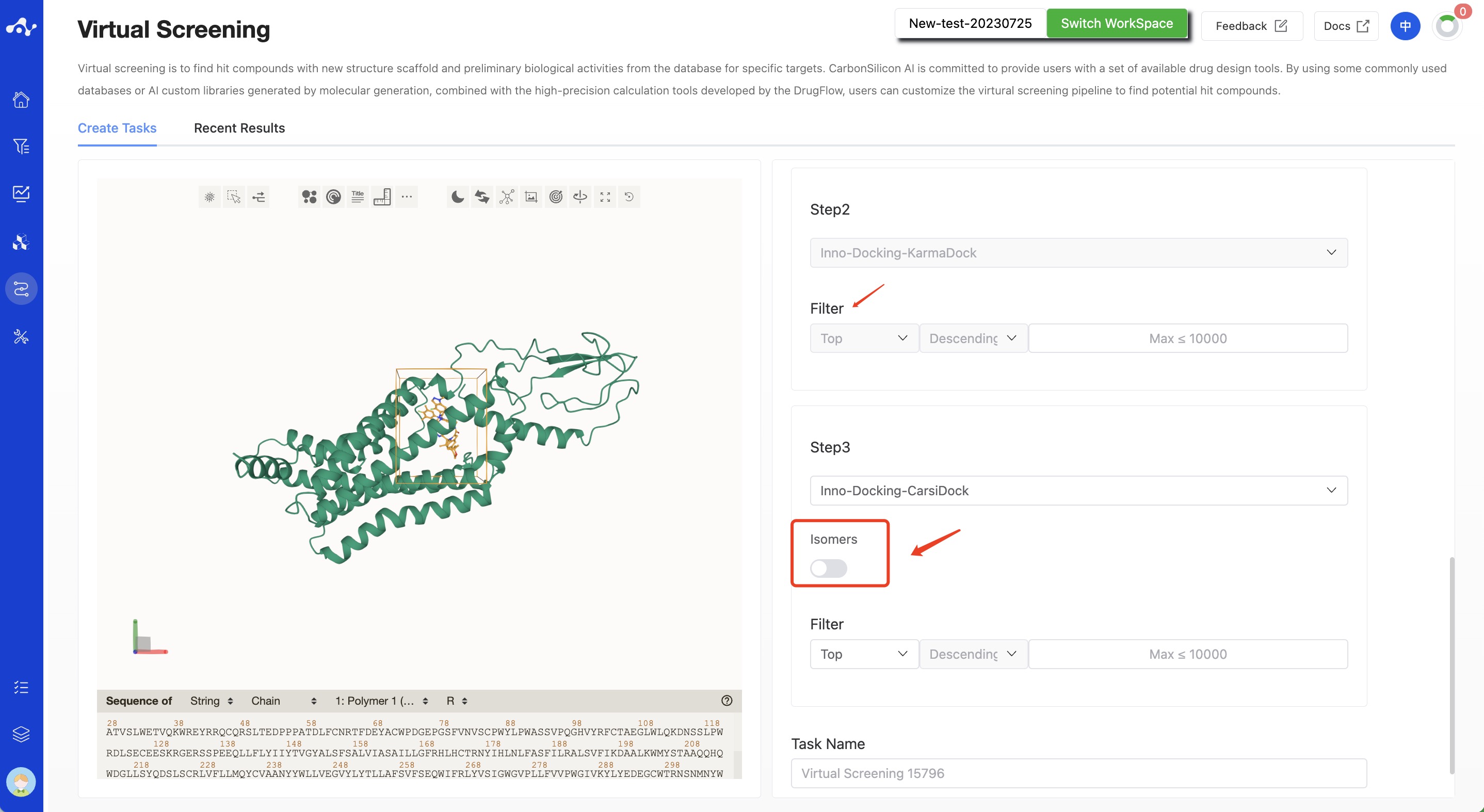
Figure 3. Setting KarmaDock and CarsiDock Filtering Parameters
(4) Running Sataus and View Results
After the task is submitted, the page will automatically jump to the "Recent Results" subpage of the current page. Here you can view the task running status of the current module (progress bar), and you can also view all running tasks of all modules in the "Running" dropdown box in the upper right corner. When the data volume is large, the system will calculate in batches, so as long as a batch of data is calculated (while the entire task is still running), you can click the "Result Details" button to enter the result page and view the prediction result list of the currently completed calculations (molecules that have not completed the calculation will not be displayed temporarily). You can also refresh the current page to get the latest completed data.
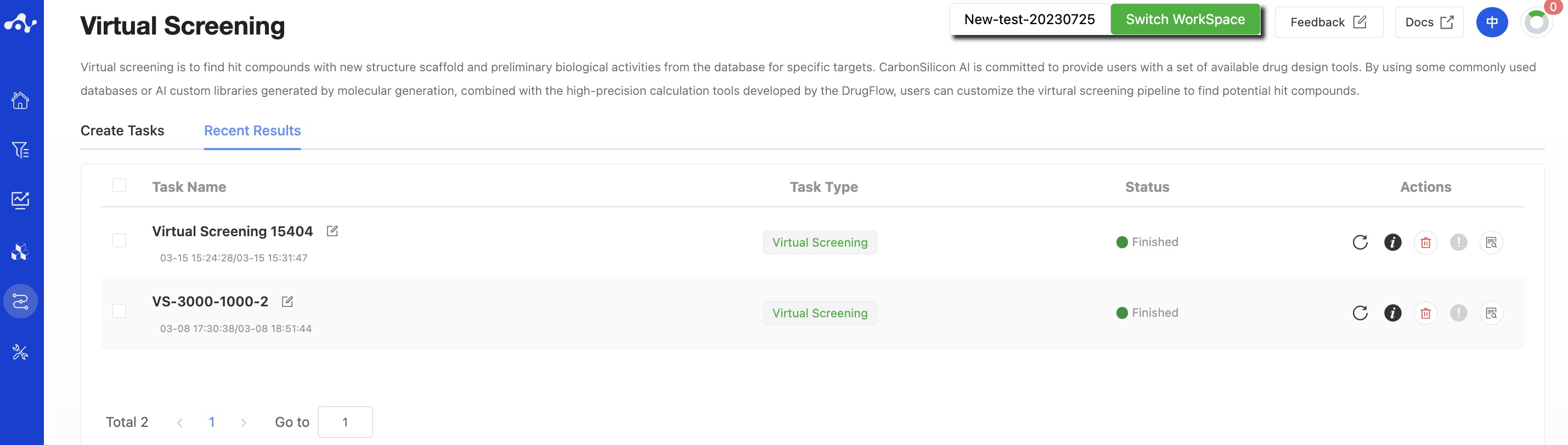
Figure 4. View Results
3. Results Analysis
The results page is composed of a Summary at the top, a molecule list on the left, and a protein visualization area on the right. By default, the result detail area on the left displays the grid page, which can be switched to the list and cluster subpages. Among them, the grid subpage provides a concise page for viewing molecule structures, while the list page provides detailed calculation results, which facilitates your analysis of the current data. The cluster subpage classifies and displays the obtained molecules according to the Murcko skeleton. The protein visualization area is a fixed content. No matter what subpage is on the left, the protein visualization area will display the protein-ligand docking mode. Users can quickly browse the interactions between molecules through the "Previous" and "Next" buttons at the bottom right corner.
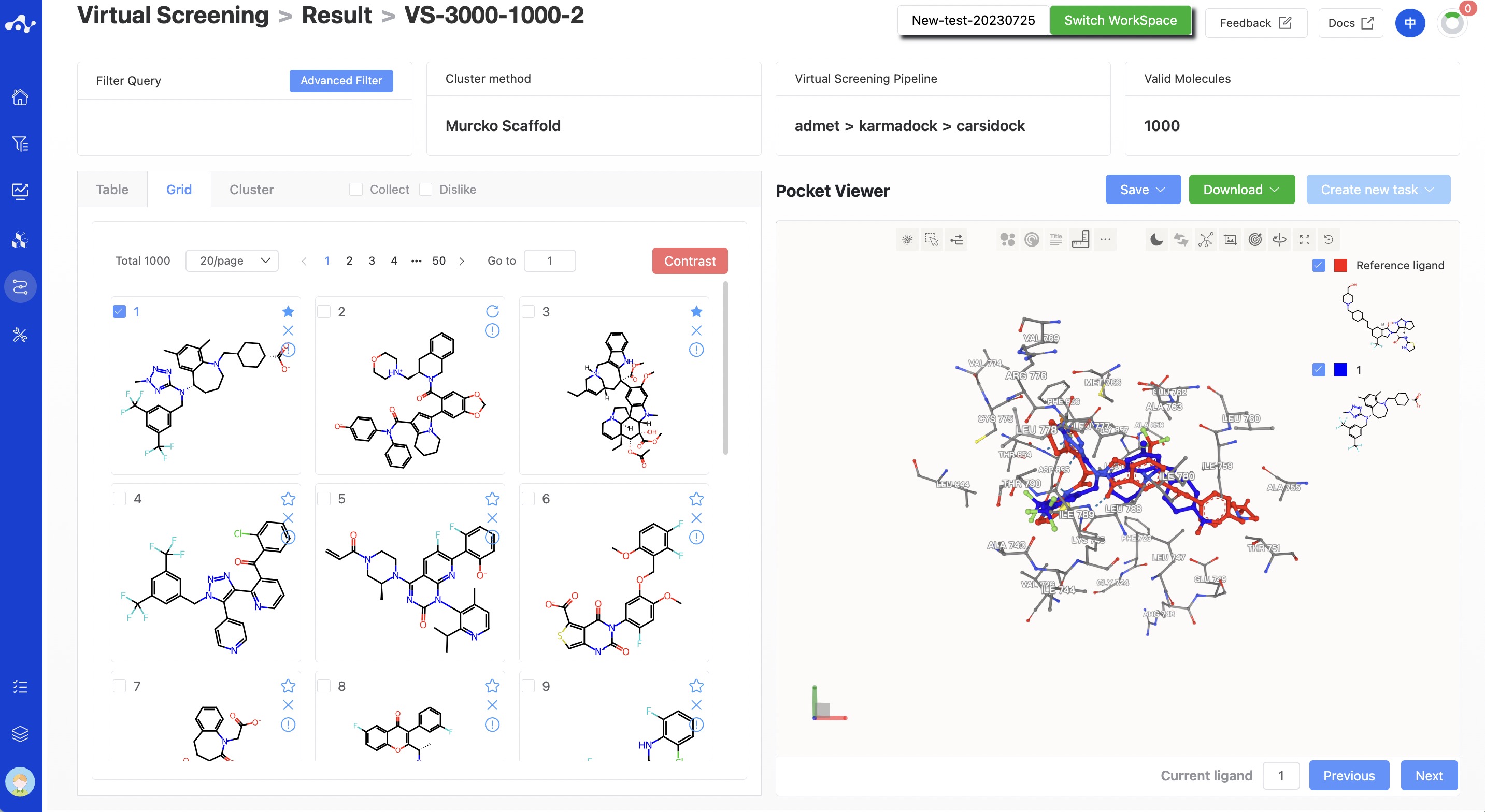
Figure 5. Virtual Screening Results Page - Default Grid Page
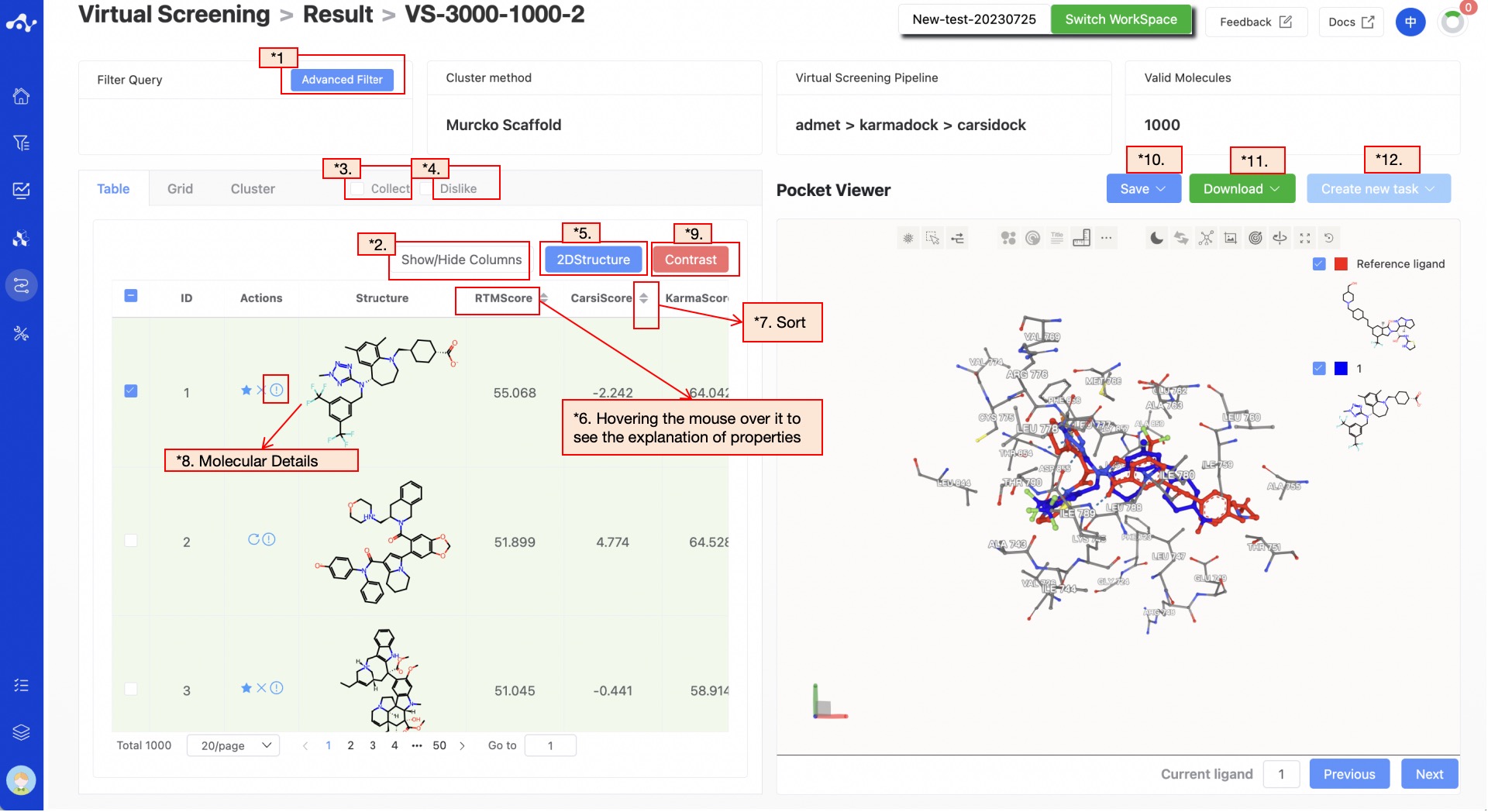
Figure 6. Virtual Screening Results Page - List Page
(1) Advanced Filtering
Advanced filtering provides range filtering, which can further filter out molecules within a specified range of certain properties to exclude molecules that do not meet the expected results. After advanced filtering, only molecules that meet the filter conditions will be displayed on the page.
(2) Show/Hide Upload Column
The default result list does not display information in the uploaded file, so it is unselected in the left control bar. When you don't want to display this property, deselect it, and the result list on the left will show in real time based on the selection in the control bar. At the top, there are also two shortcuts "Select All" and "Deselect", which are convenient for users to quickly select.
(3) Favorites
This feature is mainly used to help users mark their favorite molecules. When you click to add a molecule to your favorites, the icon will be highlighted, meaning that the molecule is marked as a favorite. After clicking the favorite checkbox, the page will only display the favorited molecules. After favoriting, you can click the chart again to cancel the favorite.
(4) Dislike
This feature is mainly used to help users mark molecules they do not like. Once a molecule is marked as disliked, the bookmark icon will disappear, and the dislike icon will be replaced with a restore icon, meaning the user can modify the label of the molecule at any time. After clicking the dislike checkbox, the page will only display molecules marked as disliked.
(5) 2D Structure
This button is selected by default, and the ligand structure is normally displayed in the list. Clicking once deselects it, and the molecule structure in the list will be hidden.
(6) Property Explanation
Hover the mouse over the name of each property to view the interpretation of the corresponding attribute.
(7) Sorting
Click the property name in the result list to reorder. For example, F(20%), click once for ascending order, click again for descending order, and click a third time to restore the original order.
(8) Molecule Detail Page
Clicking the "Molecule Details" button will take you to the molecule details page, which provides a comprehensive display of various information about the molecule, allowing users to quickly understand the molecule's detailed information, including the content of this calculation, and the Inno-ADMET properties and docking conformation of the molecule.
When you click on Inno-ADMET, you can see the physicochemical properties, ADME properties, drug-likeness, absorption, distribution, metabolism, excretion, and toxicity properties of the current molecule. Placing the mouse on the right side of each property will show its explanation and recommended range.
Clicking on docking conformation, you can see the complex conformation of the current molecule when it binds to the protein pocket. In addition, the binding conformation of the reference molecule is also provided for comparison.
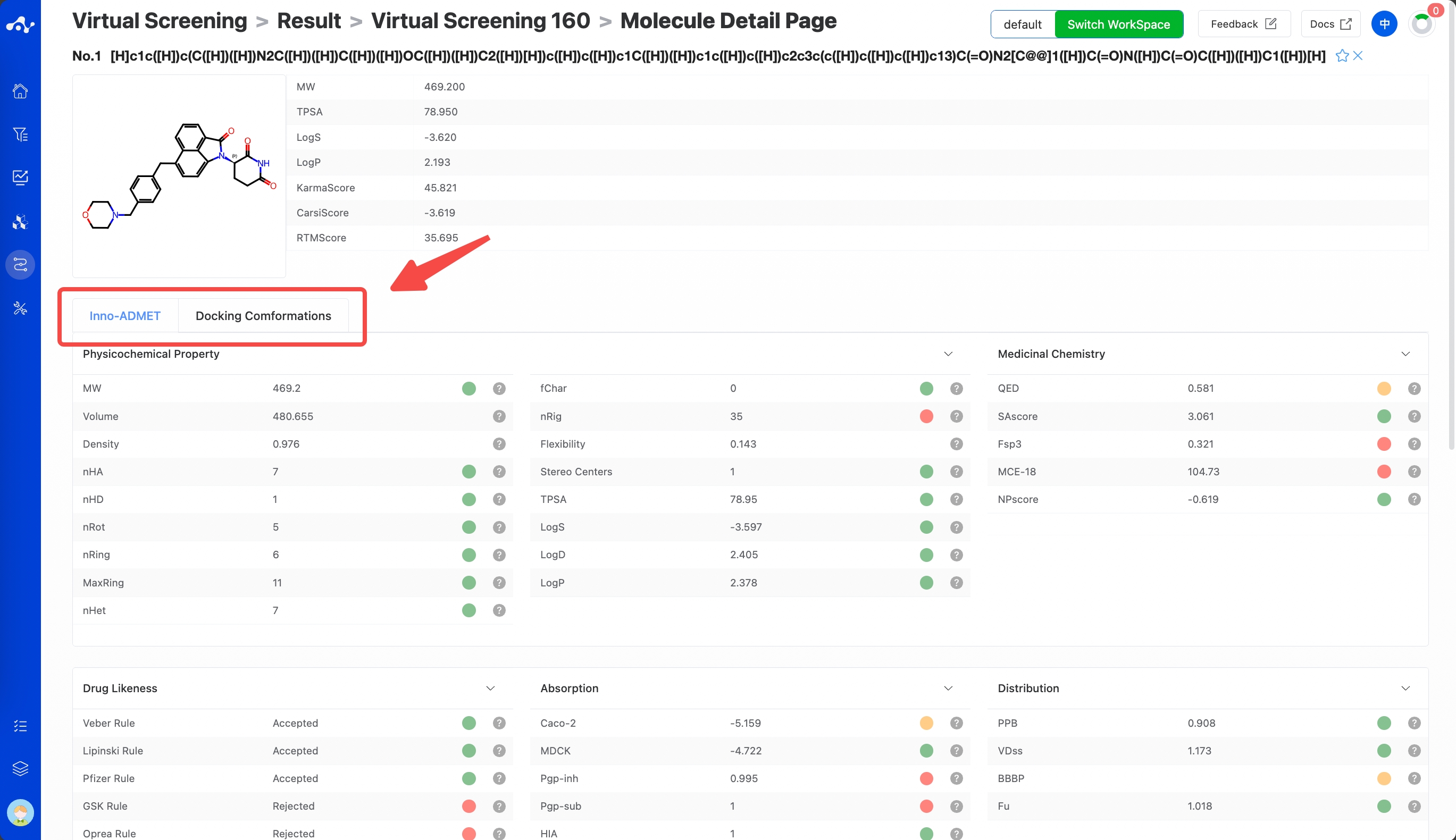
Figure 7. Virtual Screening Results Page - Molecular Details
(9) Compare
There is a checkbox at the front of each molecule's serial number. When you check multiple molecules simultaneously, you can click the [Compare] button, and the page will be directed to the compare page, which is convenient for comprehensive comparison of multiple molecules. When you have a reference molecule, it will be compared with the reference molecule. This page is similar to the single molecule detail page, containing information about the molecule's Inno-ADMET properties and docking conformation.
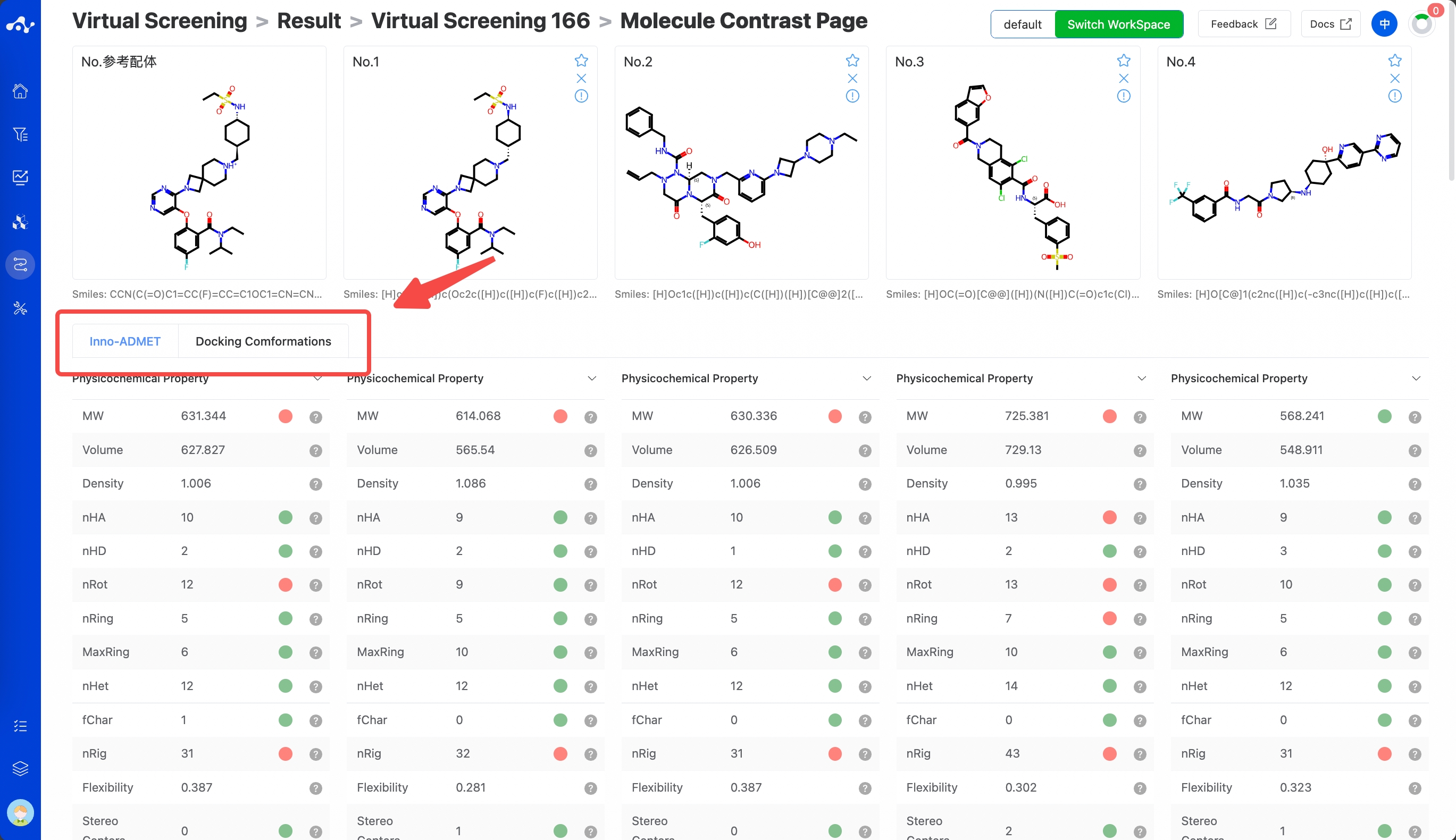
Figure 8. Virtual Screening Results Page - Molecule constrast page.
(10) Save
Click "Save", and the system will pop up a dropdown box for you to choose the file format to save (currently only supports .csv/.sdf). Once you have determined the style of the file to save, save the corresponding data to the data center as a sdf or csv file. The saved content is the molecules of the effective number displayed on the page, which are usually obtained according to your show/hide column conditions, advanced filtering conditions, favorites, or dislikes.
(11) Download
Click "Download", and the system will pop up a dropdown box for you to choose the file format to download (currently only supports .csv/.sdf). After determining the style of the file to download, the system will download the corresponding data to your local device as a sdf or csv file. The content downloaded is consistent with the save method, which also downloads the molecules of the effective number displayed on the page, which are usually obtained based on your show/hide column conditions, advanced filtering conditions, favorites, or dislikes.
(12) Create New Task
The prerequisite for creating a new task is to first save the data into a file. Before the save operation is performed, this button is disabled. As soon as the new file is saved based on the results, this button is enabled. When you click this button, the system will pop up a dropdown box for you to select the module to be calculated. After clicking, the page will immediately open a new tab and will take your saved dataset with it. After adjusting the parameters, you can submit a new task.
4. Introduction to the Related Algorithm - KarmaDock
KarmaDock is an AI docking algorithm developed by CarbonSilicon AI. It effectively combines docking and scoring methods on a lightweight model through multi-stage learning of scoring and docking, compressing the time consumption of single small molecule docking + scoring to 17ms, while ensuring docking and virtual screening performance. KarmaDock uses a model framework built with encoders (Graph Transformer (GT) and Geometric Vector Perceptrons (GVP)), and the EGNN module for docking and the mixed density network MDN module for scoring. Modeling principle: First, the protein and ligand are represented as a three-dimensional residue graph and a two-dimensional molecule graph, respectively. After encoding with the GVP/GT encoder, the pi, mu, and sigma of the mixed Gaussian distribution in the MDN module are calculated. The graph features are then fed into the EGNN module for docking to get the docking conformation, which is then sent to the MDN module for scoring to get the Score. Its modeling framework is detailed in Figure 7. KarmaDock is an end-to-end AI model that simultaneously includes docking and scoring. It achieves millisecond response on a single GPU and surpasses most docking/scoring tools in precision. It can realize high-throughput screening of large-scale molecular libraries, accelerating drug development. The average time KarmaDock takes to dock a molecule is 0.017s/complex, which is far lower than other models. KarmaDock has a docking success rate (RMSD<2 Å) of 0.956 on the Coreset dataset, surpassing most models. Moreover, KarmaDock has an enrichment factor (EF1%) of 23.4 on the Casf2016 dataset, which is at the leading level. See Table 1 for details.
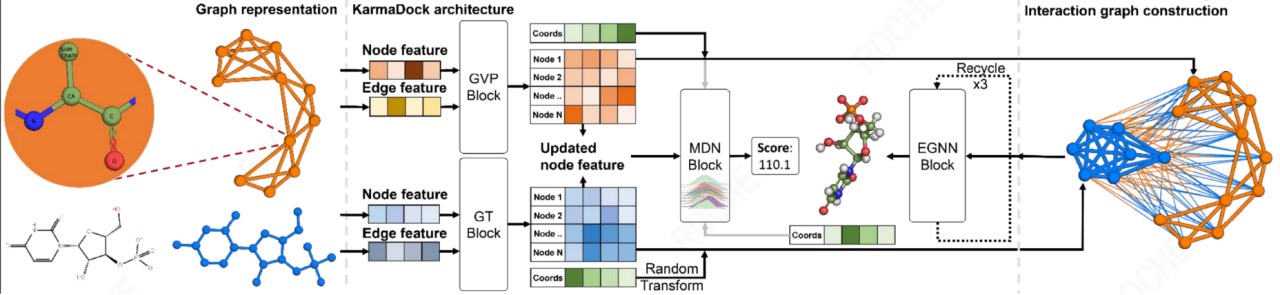
Figure 9. Modeling Framework of KarmaDock Table 1. Docking and Screening Capabilities of Various Models on CASF 2016
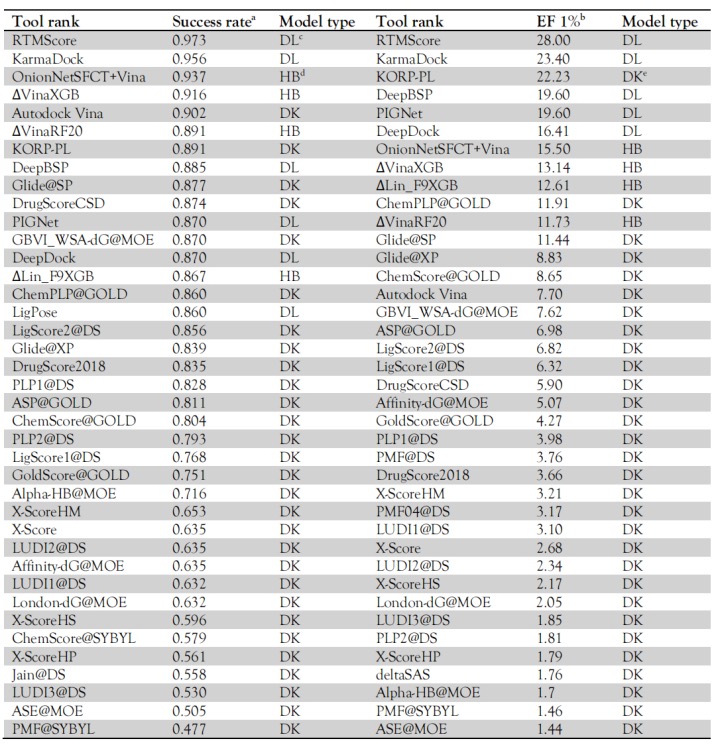
Note: For introductions to CarsiDock and RTMScore, see Inno-Docking and Inno-Rescoring, respectively.
5. Related Literature
[1] Efficient and accurate large library ligand docking with KarmaDock. Zhang X. Zhang O. Shen C., et al. Nat Comput Sci. 2023 Sep;3(9):789-804. doi: 10.1038/s43588-023-00511-5.