Molecular Factory
1. Overview of Molecular Factory
The Molecule Factory is a comprehensive molecular design method.
Designing novel and therapeutic drug-like molecules has always been at the core of new drug development. Compared to humans, AI molecule generation models, represented by deep learning, have the ability to learn from vast amounts of data. They are capable of exploring a broader chemical space and possess specific molecule optimization capacity for particular molecular properties. Current AI molecule generation techniques, such as scaffold-hopping, fragment generation, and de novo design, have been widely used in many critical molecule design and optimization projects. They navigate the chemical space more effectively, letting researchers focus on a smaller range of molecules specific to their target. Furthermore, after generating a large number of molecules for a given target and property constraints using AI, building a comprehensive and efficient automated screening process — based on molecular physicochemical properties, drug-likeness, key interplays of molecule and target, and expert insight — will help to select potentially effective molecules, thus improving R&D efficiency.
By efficiently integrating AI molecule generation techniques and automated screening processes, we have constructed the Molecule Factory module to assist researchers and practitioners in conducting efficient drug research and development procedures.
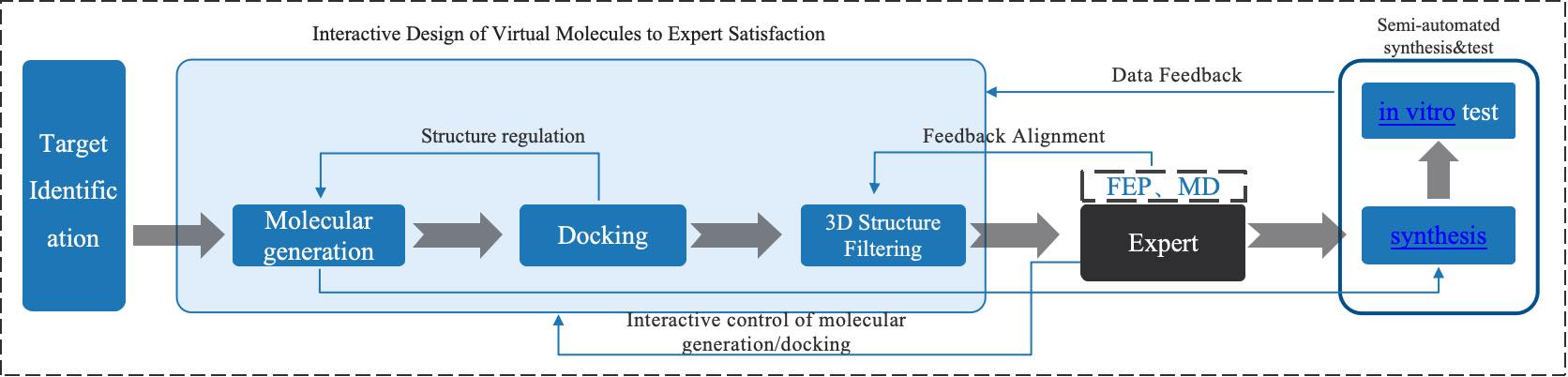
Figure 1. The Core Design Concept of the Molecular Factory.
After analyzing a specific protein target to determine the molecular generation plan and screening conditions, users can utilize the Molecule Factory module to automatically complete the entire process, including gaining molecules for screening based on the generation model and screening those molecules to obtain potential active ones based on given conditions. The core features of the Molecule Factory include:
(1) Highly automated molecular design and screening procedure: The Molecule Factory covers classic drug molecule design processes including molecule generation, physicochemical property screening, drug-likeness screening, molecule docking, and screening for key protein-ligand interactions. Traditionally, screening for key protein-ligand interactions is laborious and time-consuming, often done manually. The Molecule Factory automates this step, conducting batch interaction analyses on the complex conformations obtained from molecule docking, and performs automatic filtering based on expert-specified key substructures and interactions.
(2) Comprehensive functionality of molecular generation models: The molecular generation models support both ligand-based and structure-based drug design, giving users the freedom to choose whether to upload the protein structure. The molecule design modes include R-group design, linker design, scaffold-hopping, and de novo drug design, catering to a variety of design needs. Not only that, the Molecule Factory also supports users in uploading known active molecule datasets for a target, allowing the molecular generation model to be fine-tuned for more specific molecule generation for a particular target.
(3) Application of CarsiDock, the most accurate molecular docking model among all current semi-flexible docking methods: The Molecule Factory integrates CarbonSilicon AI's independently developed AI docking model CarsiDock, which uses massive complex structure simulation data produced by physical calculation to predict the binding conformation of ligand molecules and proteins quickly and accurately.
(4) Screening conditions consistent with expert experience: The Molecule Factory models the molecular design experience of experts, automatically filtering candidate molecules obtained by the molecular generation model for experience-based factors, including unreasonable substructures, molecular weight, topological polar surface area, lipophilicity, solubility, PAINS, Alerts, quantitative estimation of drug-likeness, number of hydrogen bond donors, number of hydrogen bond acceptors, synthesizability, number of rotatable bonds, number of rings, number of helices, number of bridge rings, and number of macro rings. Unreasonable structures are uncommon substructures in drug-like molecules, encompassing various categories such as substructures with low stability, rare substructures, and those with high synthetic difficulty. These all come from the expertise of veteran medicinal chemists.
(5) Significant improvement in development efficiency: Drug design and screening based on the Molecule Factory can complete a round of molecule optimization within a week, reducing early development time by 50% and early development cost by 70%.
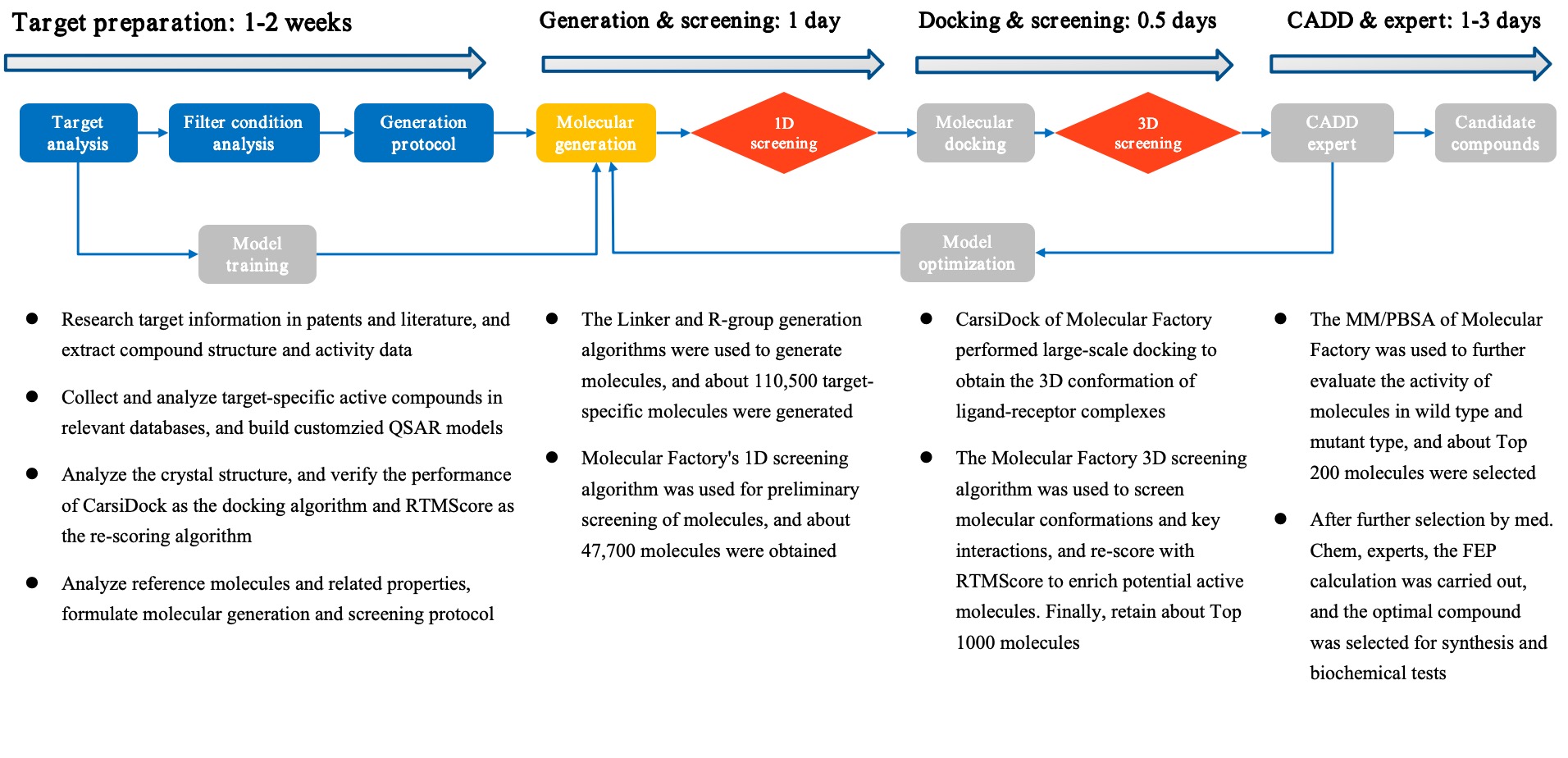
Figure 2. The Process and Time Consumption of Drug Design Aided by the Molecular Factory.
2. Instructions for Use
To complete the full process of automatic molecular design, users need to set the following sequentially: Select generation type - Upload protein and set pocket(Optional field) - Upload and edit Reference molecule(Mandatory field) - Upload active molecules(Optional field) - Setting generation method and termination conditiontask(Mandatory field) - 2D filtering(Optional field) - Docking(Optional field) - naming(Optional field) - task submission.
(1) Generation Type
Currently, the platform supports five types of molecular generation, which are:
R-Group: Replace one R group based on a reference molecule.
Sidechain-base: Replace multiple R groups based on a reference molecule.
Linker-based: Replace the middle Linker while preserving the structure at both ends, based on a reference molecule.
Scaffold-hopping: Replace the central scaffold while preserving multiple structures at the ends, based on a reference molecule.
De Novo: Generate from scratch.
(2) Upload protein and set pocket (Optional field)
If the user does not need to base molecular generation on the protein structure, this step can be skipped; otherwise, if the user wants to generate molecules based on the protein structure, then they need to upload the protein and set the pocket.
- Protein Upload
The platform provides two methods of protein upload: file upload and data center. After the protein is uploaded, it will automatically be visualized on the left.
Upload file: Check the "Upload file" checkbox, and select the local file by clicking the button below. The currently supported file format is .pdb. After the file is selected, its name will be displayed on the button;
Data center: Select the "Data center" checkbox, and a popup will appear when you click the button below. You can select the data center's data by clicking on the file name, and the popup will disappear after clicking.
- Pocket Setting
Specifying the binding pocket of the molecule, there are two ways to do this: Based on the PDB file you uploaded, the system will automatically select the molecule with the largest molecular weight as the pocket, and determine the geometric center coordinates and Box Size based on this ligand molecule; you can also customize the pocket by manually adjusting the coordinates and Box Size of the pocket.
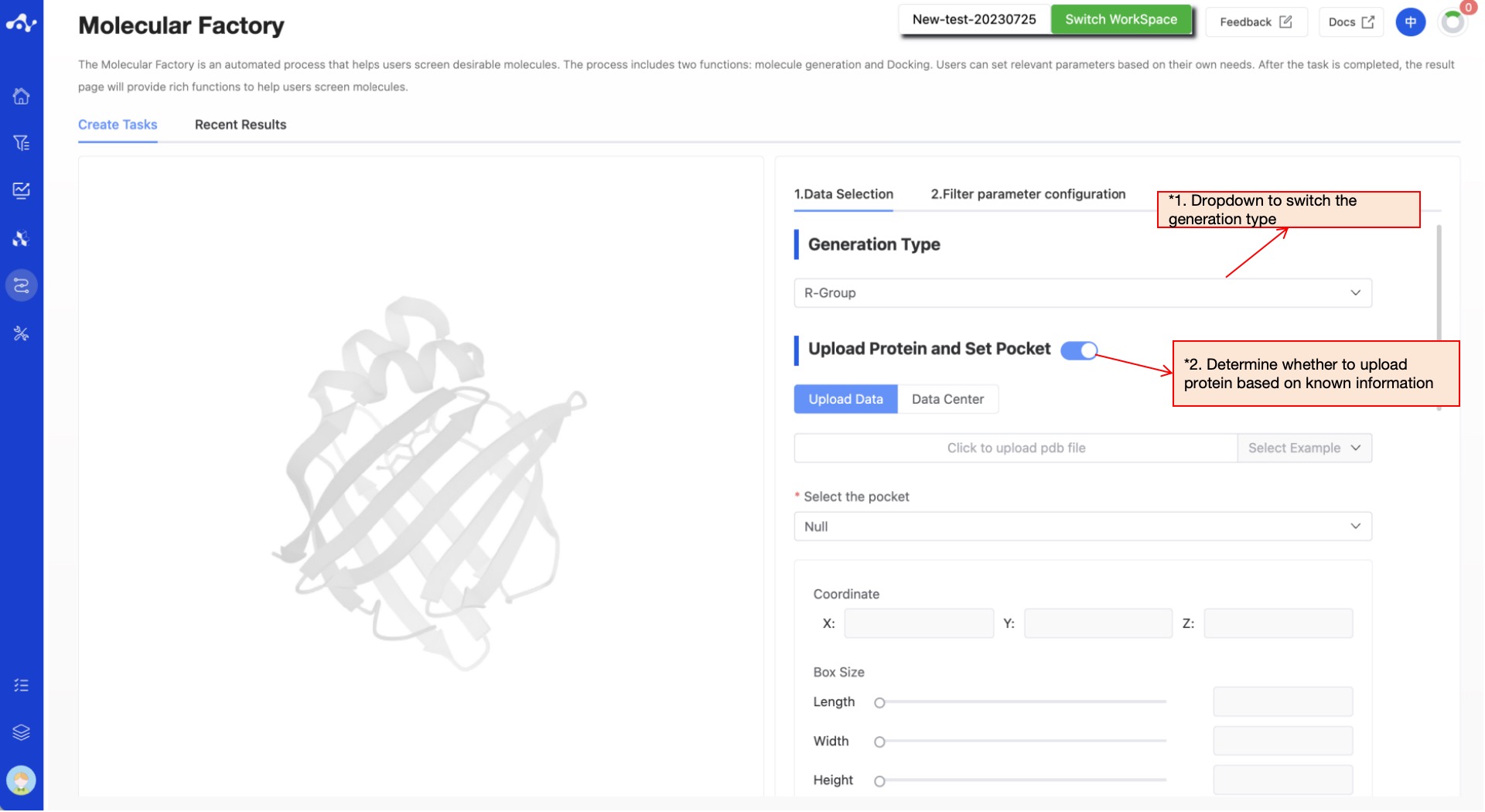
Figure 3. The page of create Molecular Factory task - upload proteins and set pockets
(3) Upload and edit Reference molecule(Mandatory field)
The platform supports four ways of uploading: using the complex ligand, entering SMILES, upload file and select from the data center.
Regarding the complex ligand: If you are doing molecular generation based on the protein pocket, when the pdb file you uploaded contains the complex ligand, the system will prioritize extracting and drawing the ligand of the complex. Sometimes, there can be cases where extraction of the complex ligand fails (the front-end page will provide relevant prompts), in such cases, you can upload the ligand of the complex as a reference molecule using the other three methods, or upload another molecule as the reference molecule. The system will re-dock this molecule into the specified pocket, and then perform molecular generation based on your specified site.
Regarding location-specific: Use the molecular editor to select and delete the substructure expected to be replaced from the uploaded reference molecule/reference fragment. The platform will automatically recognize the starting atom position for molecular growth based on the editing results.
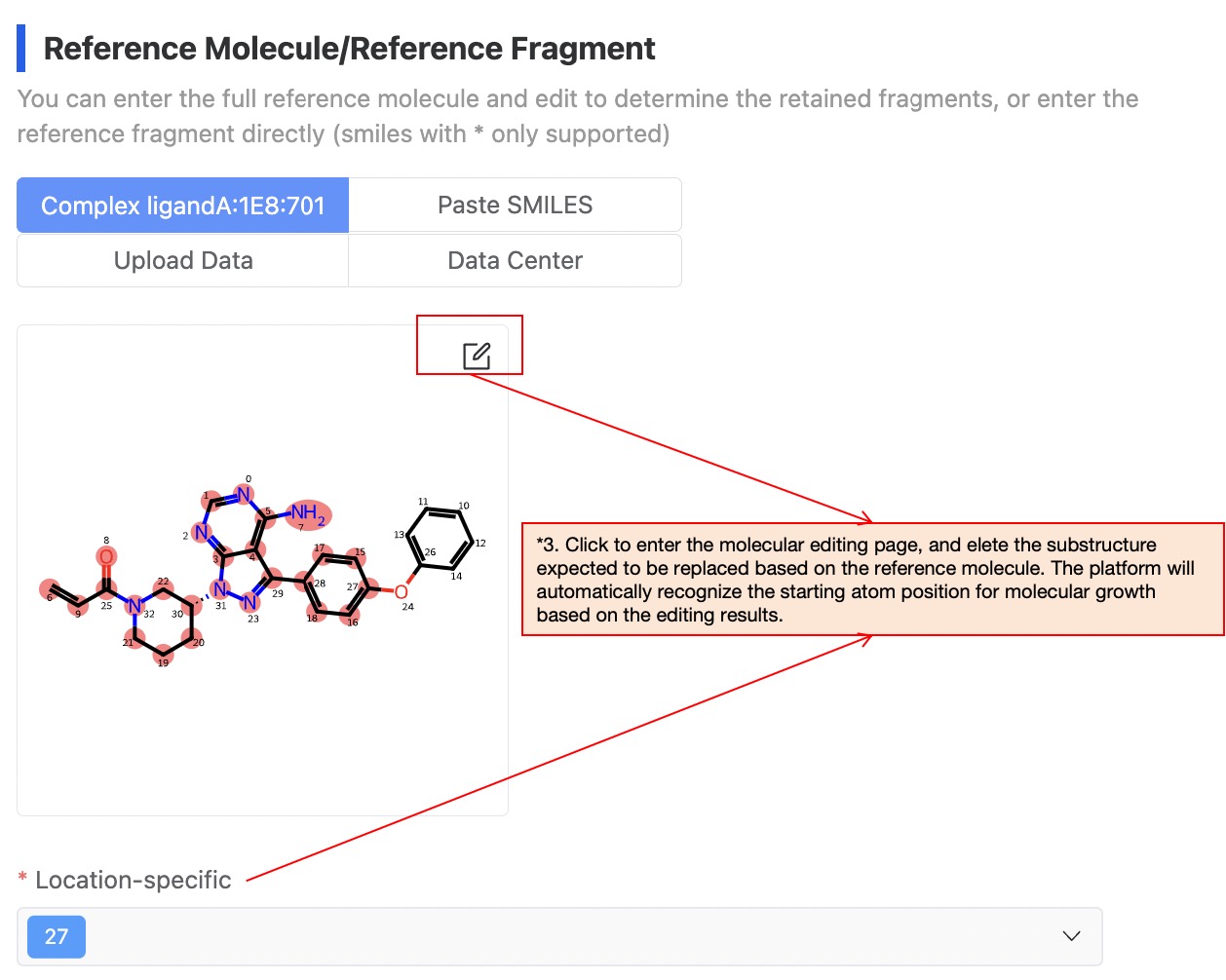
Figure 4. Upload and edit Reference molecule
(4) Upload active molecules(Optional field)
If you have collected a batch of active molecules corresponding to the target, they can upload these active molecules. The platform will automatically fine-tune the generation model based on these active molecules, and generate molecules based on the fine-tuned model, so that the generated molecules are more structurally/property similar to the active molecules. If not necessary, this step can also be skipped.
- Upload File
Check the "Upload File" box, and you can select a local file by clicking the button below. After the file is selected, the file name will be displayed on the button, and the file content will be displayed on the right. Regarding the uploaded files:
The currently supported file formats are .sdf and .csv. If an sdf file is uploaded, the task can be directly submitted. If a csv file is uploaded, you need to specify the smiles column before the task can be submitted.
The file size should not exceed 10MB.
To ensure good fine-tuning results, the number of active molecules uploaded should not be less than 300.
- Data Center
Check the "Data Center" box, and a pop-up window will appear when you click the button below. Click on the file name to select data from the data center. After you click, the pop-up window will disappear and you can submit the task.
(5) Setting generation method and termination conditiontask(Mandatory field)
The platform provides several molecular generation methods: ResGen, CarsiLinker, CarsiLinker3D, FragGPT, Delete, Smiles-GPT. Each method supports different molecular generation modes, and the system will automatically determine the generation method based on your input. You can deselect any of the available algorithms if needed. Set the generation quantity and generation time. Once either of these conditions is met, the platform will automatically terminate the generation task.
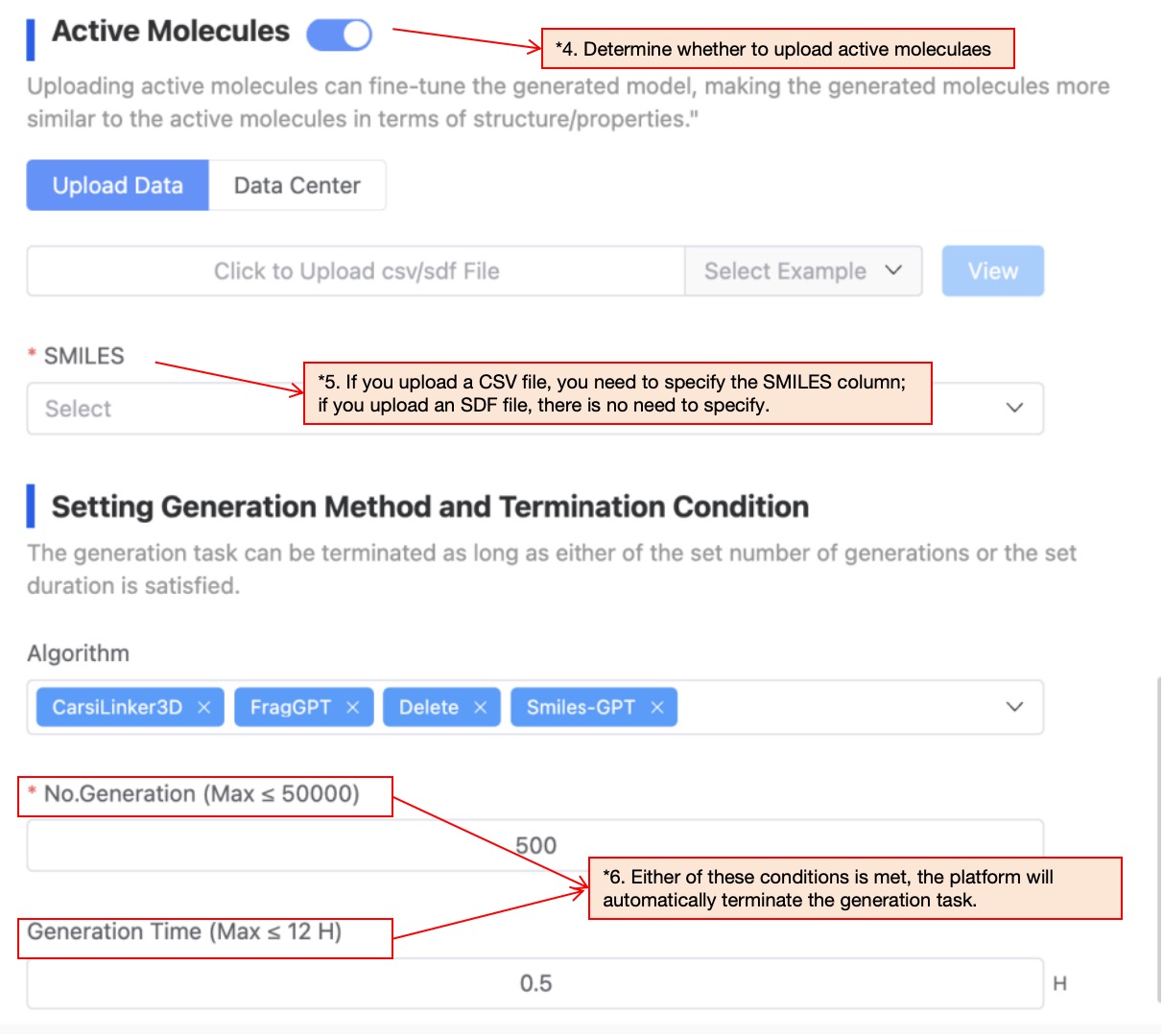
Figure 5. Upload active molecules and set generation method/termination conditions After determining all parameters, click 'Next' to proceed to the filter parameter configuration.
(6) 2D filtering(Optional field) - Docking(Optional field) - naming(Optional field) - task submission.
If users do not wish to filter the generated molecules by 2D properties, they can skip this step; if they wish to filter the generated molecules by 2D properties, continue to select the type of property and adjust the target range of the property. The platform supports 4 types of properties, including MW, TPSA, LogP, and LogS. 
Figure 6. Filter condition configuration — 2D property filtering
(6) Docking(Optional field)
This step is used for automatic docking of the generated molecules to generate complex conformations. Users can choose to skip this step if they do not need it. If needed, continue to configure the generation parameter of isomers one by one, and choose one docking method from CarsiDock and AutoDock Vina.
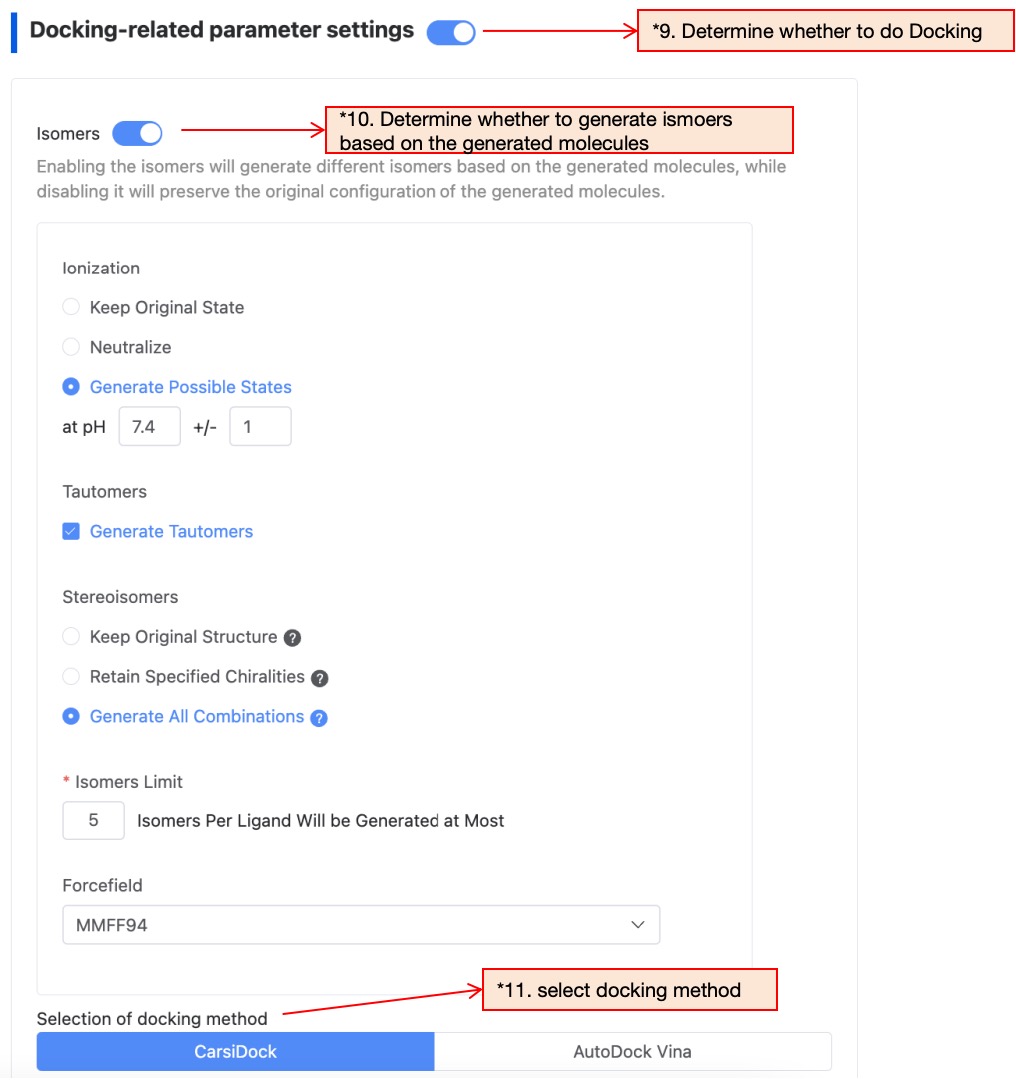
"Figure 7. Filter condition configuration — Docking related parameter settings
- Docking conformation comparison (optional)
This step is used to compare the 3D conformation differences between the common substructures when the generated molecule and the reference molecule bind to the protein. If you do not need this step, you can turn it off.
- Atom spatial position detection (optional)
This step is used to specify key atoms in the reference molecule, to detect whether there are atoms of the same type in the generated molecule within a 5 angstrom range of the specified atom in the complex conformation. If you do not need this step, you can turn it off. If needed, select the atom from the drop-down menu."
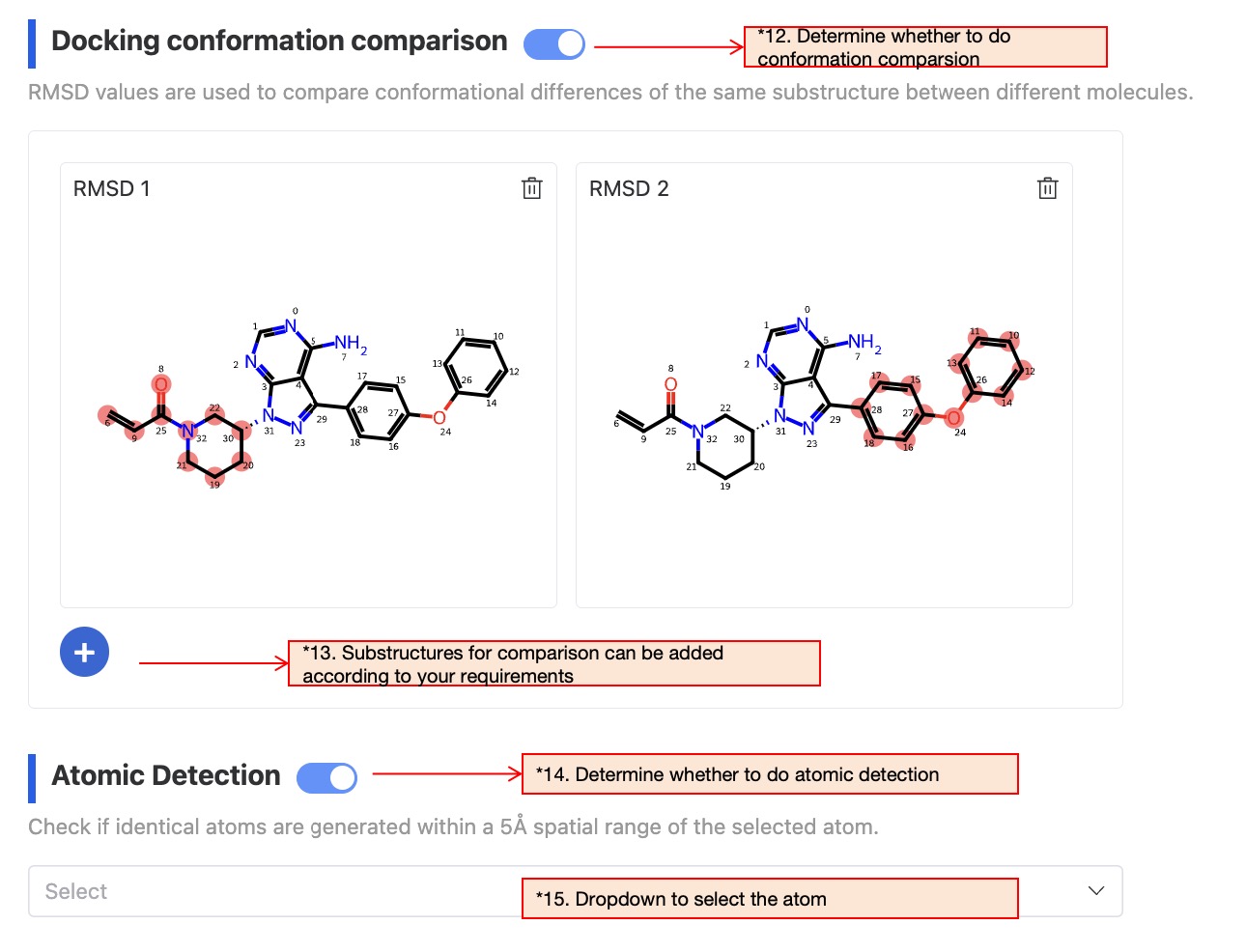
Figure 8: Docking conformation comparison and atom spatial position detection After all parameters are set, name the task and click 'Submit' to complete the task submission.
(3) Running Sataus and View Results
After the task is submitted, the page will automatically jump to the "Recent Results" subpage of the current page. Here you can view the task running status of the current module (progress bar), and you can also view all running tasks of all modules in the "Running" dropdown box in the upper right corner. When the data volume is large, the system will calculate in batches, so as long as a batch of data is calculated (while the entire task is still running), you can click the "Result Details" button to enter the result page and view the prediction result list of the currently completed calculations (molecules that have not completed the calculation will not be displayed temporarily). You can also refresh the current page to get the latest completed data.
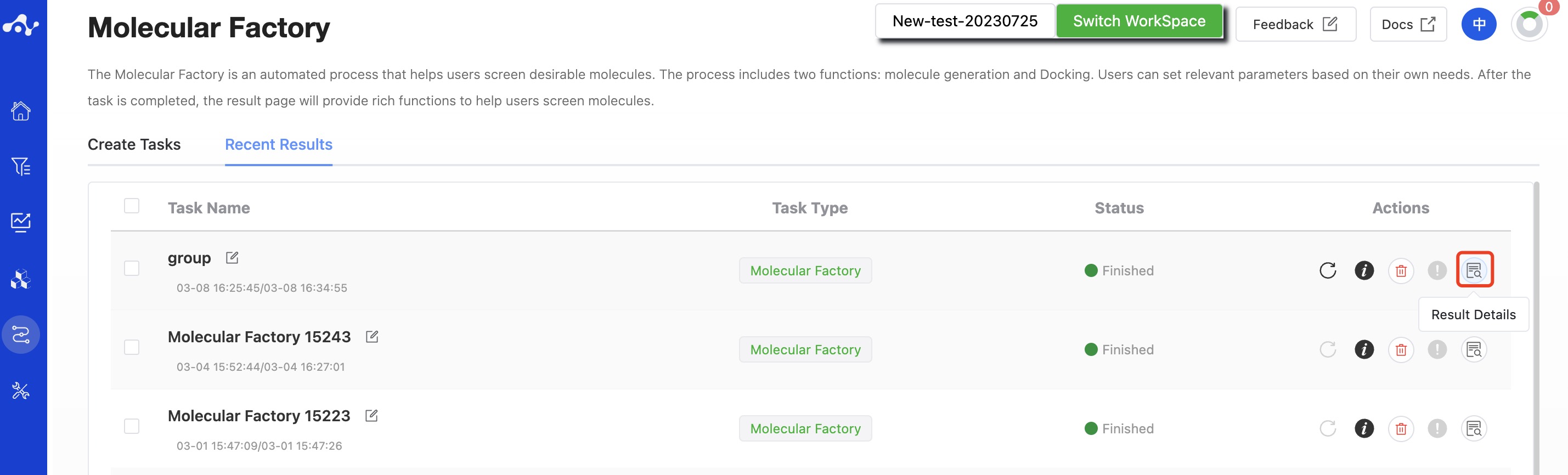
Figure 9. View results
3. Results Analysis
The results page is made up of a Summary at the top, a molecule list on the left, and a protein visualization area on the right. By default, the results detail area on the left displays the grid page, which you can switch to the list and clustering subpages. The grid subpage provides a streamlined page for viewing molecular structures, while the list page provides detailed computation results to help you analyze the current data. The clustering subpage displays the molecules categorized according to the Murcko scaffolds. The protein visualization area is a fixed content, no matter what subpage is on the left, the protein visualization area will display the protein-ligand docking mode, and users can quickly browse the interactions between molecules through the "Previous" and "Next" buttons at the bottom right.
If you did not choose to perform docking in your task, the results page will only display the generated molecules and calculated 2D properties, and there will not be a protein visualization area. On the grid page, each molecular card displays the structure of the molecule. The three symbols in the upper right corner of the card correspond to three operations, namely bookmark, dislike, and result details. Users can bookmark and dislike to mark the molecules. Clicking on the result details button will redirect to the molecule details page.
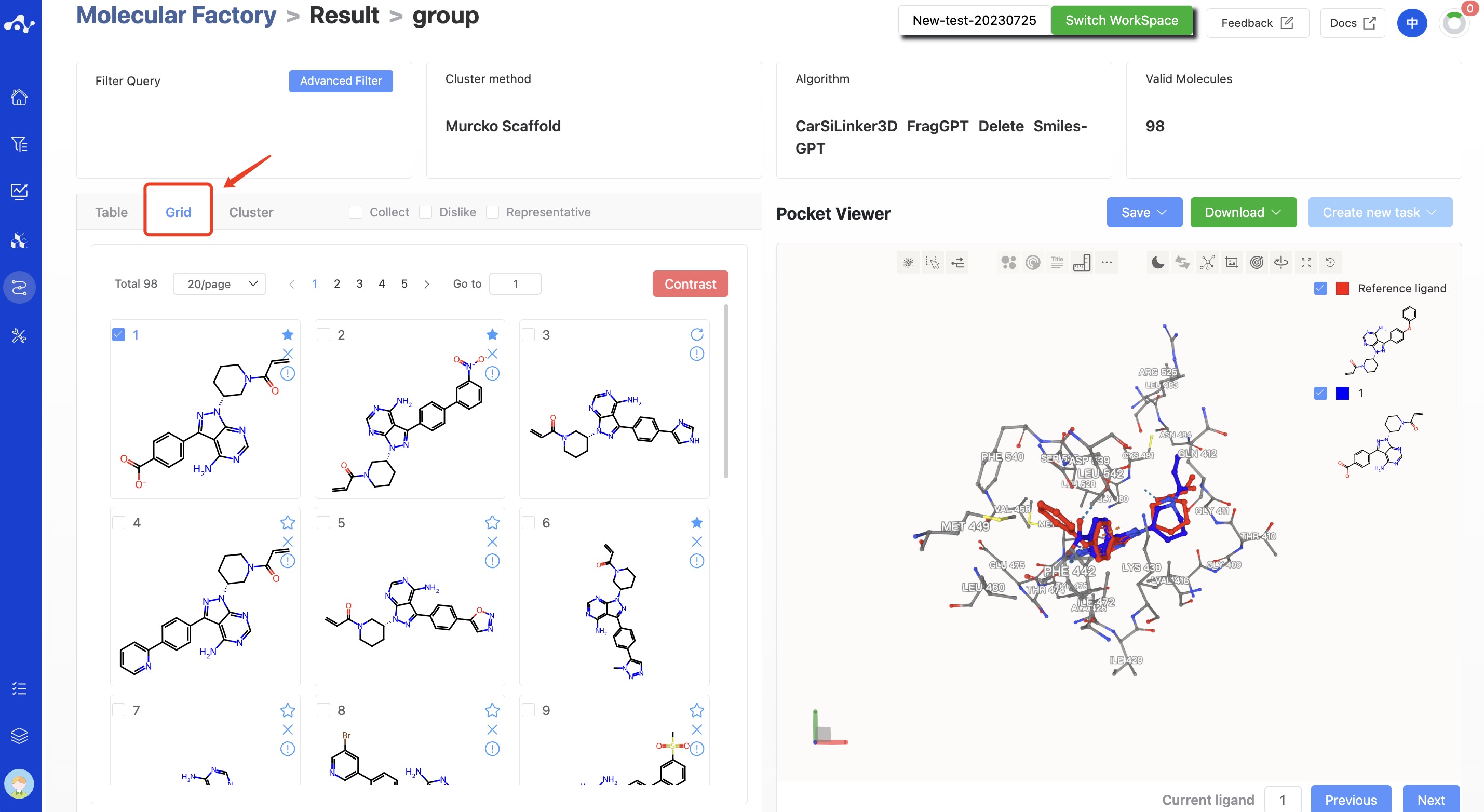
Figure 10. Molecule Factory Results Page - Grid Page
On the list page, you can view detailed information about the molecules, including the ID of the generated molecule, operations, structure, and multiple properties. Moving the mouse over the name of each property will display an explanation of that property. Clicking on the category names in the list will reorder them: clicking once will sort in ascending order, clicking again will sort in descending order, and clicking a third time will restore the original order.
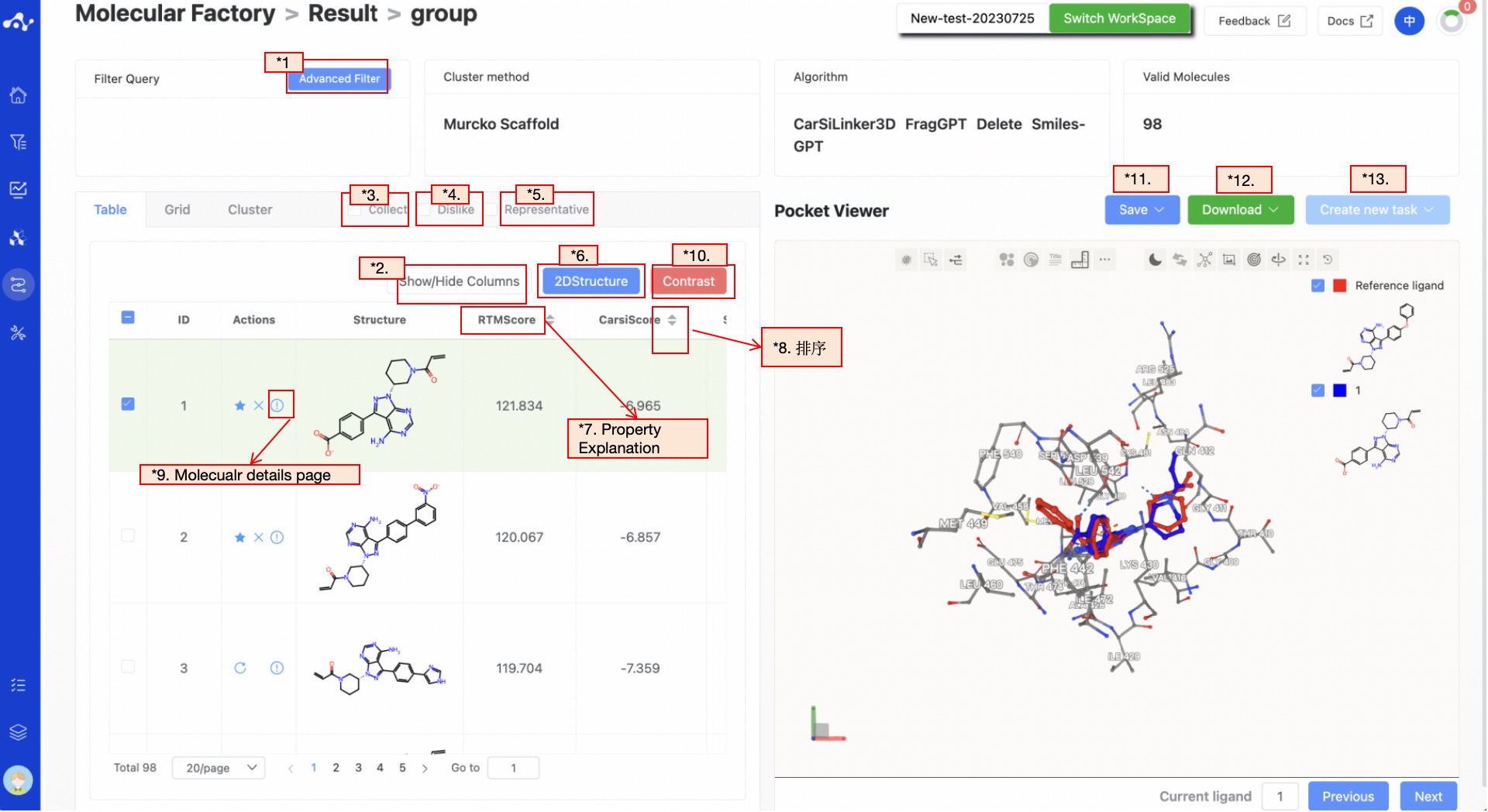
Figure 11. Molecule Factory Results Page - List Page
On the clustering page, the generated molecules are displayed in clusters according to their Murcko Scaffold, and molecules with the same scaffold will be displayed together.
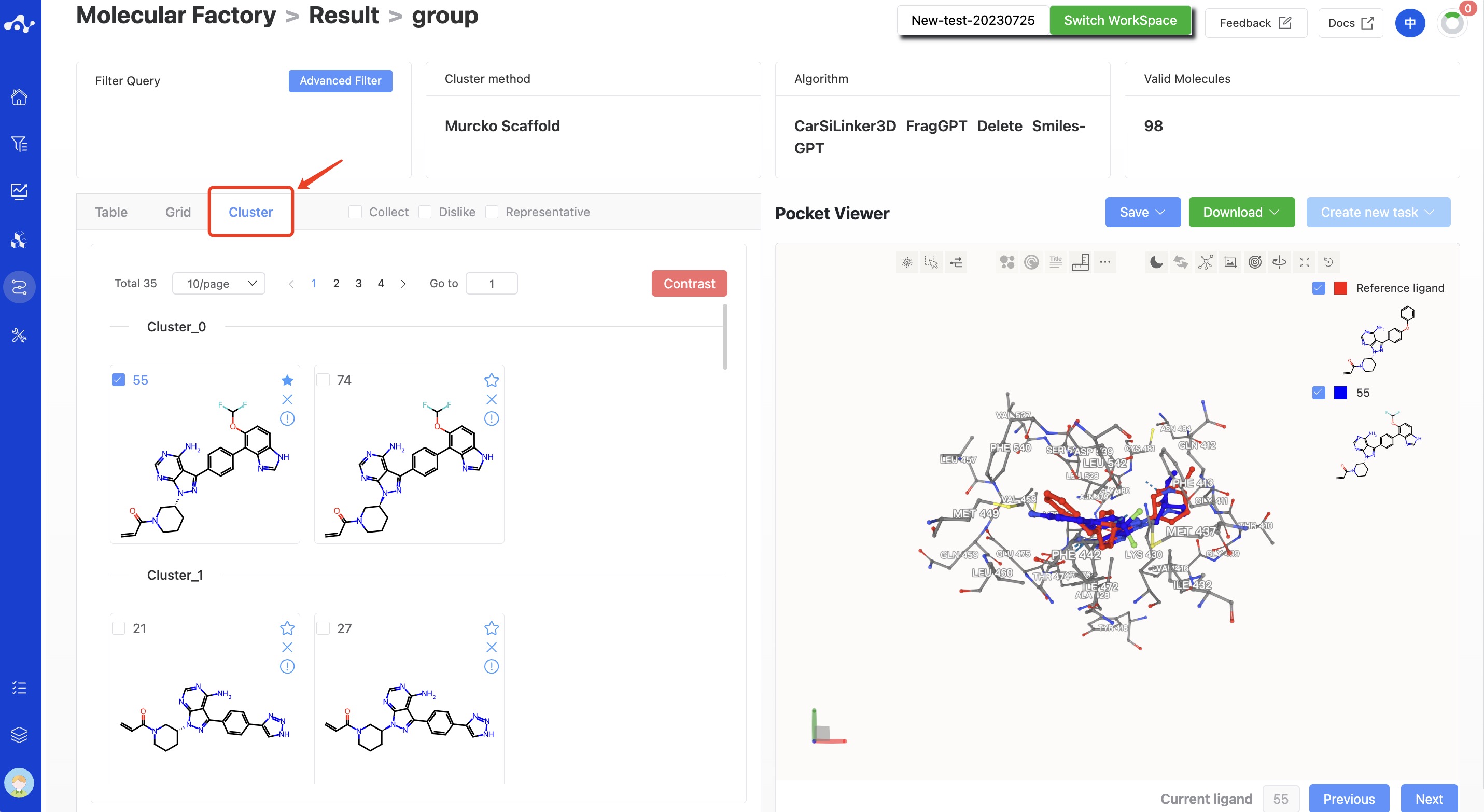
Figure 12. Molecule Factory Results Page - Cluster Page
(1) Advanced Filtering
Advanced filtering provides range filtering, which can further filter out molecules within a specified range of certain properties to exclude molecules that do not meet the expected results. After advanced filtering, only molecules that meet the filter conditions will be displayed on the page.
(2) Show/Hide Upload Column
The default result list does not display information in the uploaded file, so it is unselected in the left control bar. When you don't want to display this property, deselect it, and the result list on the left will show in real time based on the selection in the control bar. At the top, there are also two shortcuts "Select All" and "Deselect", which are convenient for users to quickly select.
(3) Favorites
This feature is mainly used to help users mark their favorite molecules. When you click to add a molecule to your favorites, the icon will be highlighted, meaning that the molecule is marked as a favorite. After clicking the favorite checkbox, the page will only display the favorited molecules. After favoriting, you can click the chart again to cancel the favorite.
(4) Dislike
This feature is mainly used to help users mark molecules they do not like. Once a molecule is marked as disliked, the bookmark icon will disappear, and the dislike icon will be replaced with a restore icon, meaning the user can modify the label of the molecule at any time. After clicking the dislike checkbox, the page will only display molecules marked as disliked.
(5) Representative
To the right of the list, grid, and cluster options, there are three additional options: bookmark, dislike, and representative. Clicking bookmark will display only the molecules marked as bookmarked in the results detail area. Clicking Representative will only display the most representative molecule in each category after clustering.
(6) 2D Structure
This button is selected by default, and the ligand structure is normally displayed in the list. Clicking once deselects it, and the molecule structure in the list will be hidden.
(7) Property Explanation
Hover the mouse over the name of each property to view the interpretation of the corresponding attribute.
(8) Sorting
Click the property name in the result list to reorder. For example, F(20%), click once for ascending order, click again for descending order, and click a third time to restore the original order.
(9) Molecule Detail Page
Clicking the "Molecule Details" button will take you to the molecule details page, which provides a comprehensive display of various information about the molecule, allowing users to quickly understand the molecule's detailed information, including the content of this calculation, and the Inno-ADMET properties and docking conformation of the molecule.
When you click on Inno-ADMET, you can see the physicochemical properties, ADME properties, drug-likeness, absorption, distribution, metabolism, excretion, and toxicity properties of the current molecule. Placing the mouse on the right side of each property will show its explanation and recommended range.
Clicking on docking conformation, you can see the complex conformation of the current molecule when it binds to the protein pocket. In addition, the binding conformation of the reference molecule is also provided for comparison.
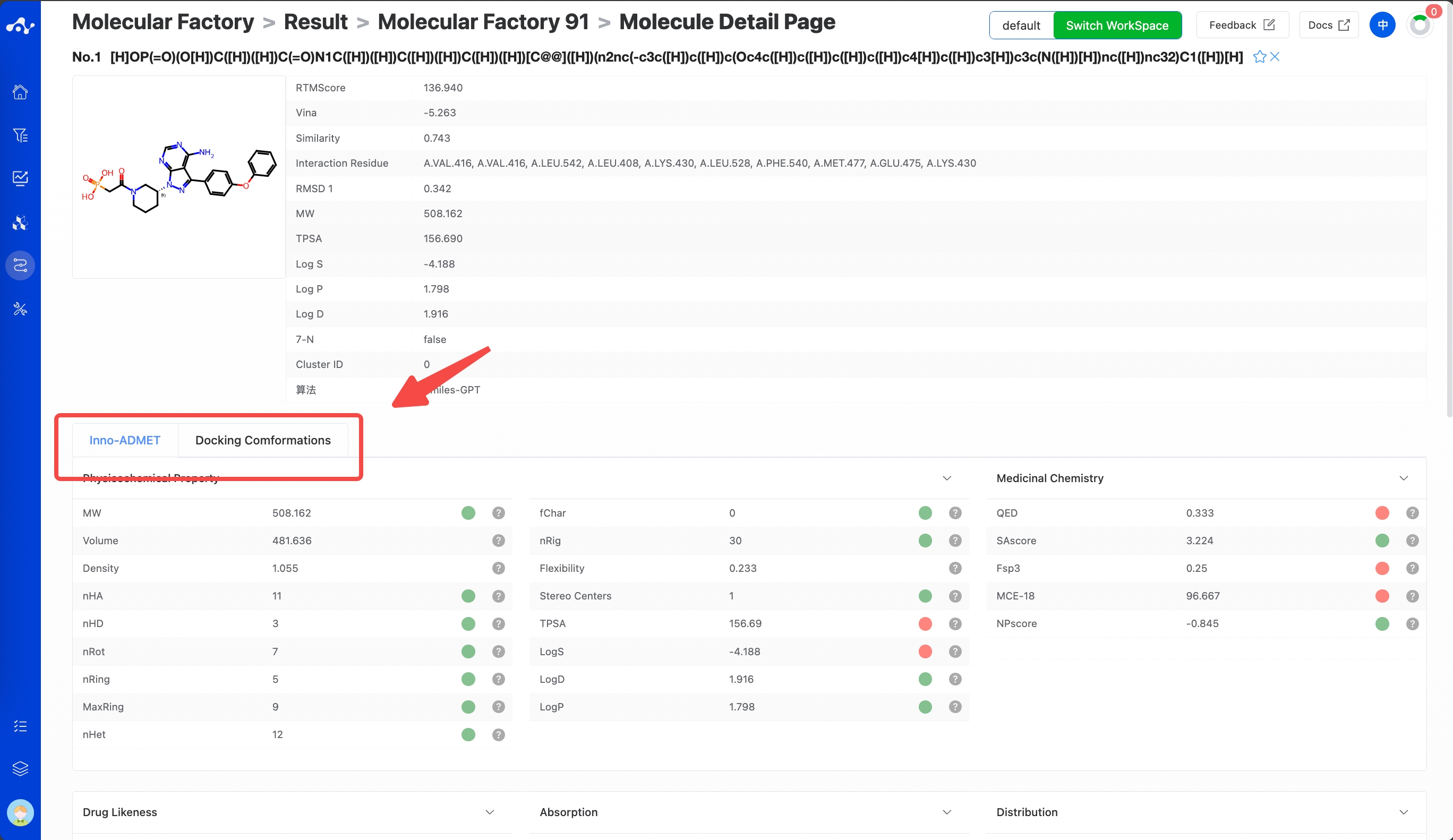
Figure 13. Single Molecule Detail Page
(10) Compare
There is a checkbox at the front of each molecule's serial number. When you check multiple molecules simultaneously, you can click the [Compare] button, and the page will be directed to the compare page, which is convenient for comprehensive comparison of multiple molecules. When you have a reference molecule, it will be compared with the reference molecule. This page is similar to the single molecule detail page, containing information about the molecule's Inno-ADMET properties and docking conformation.
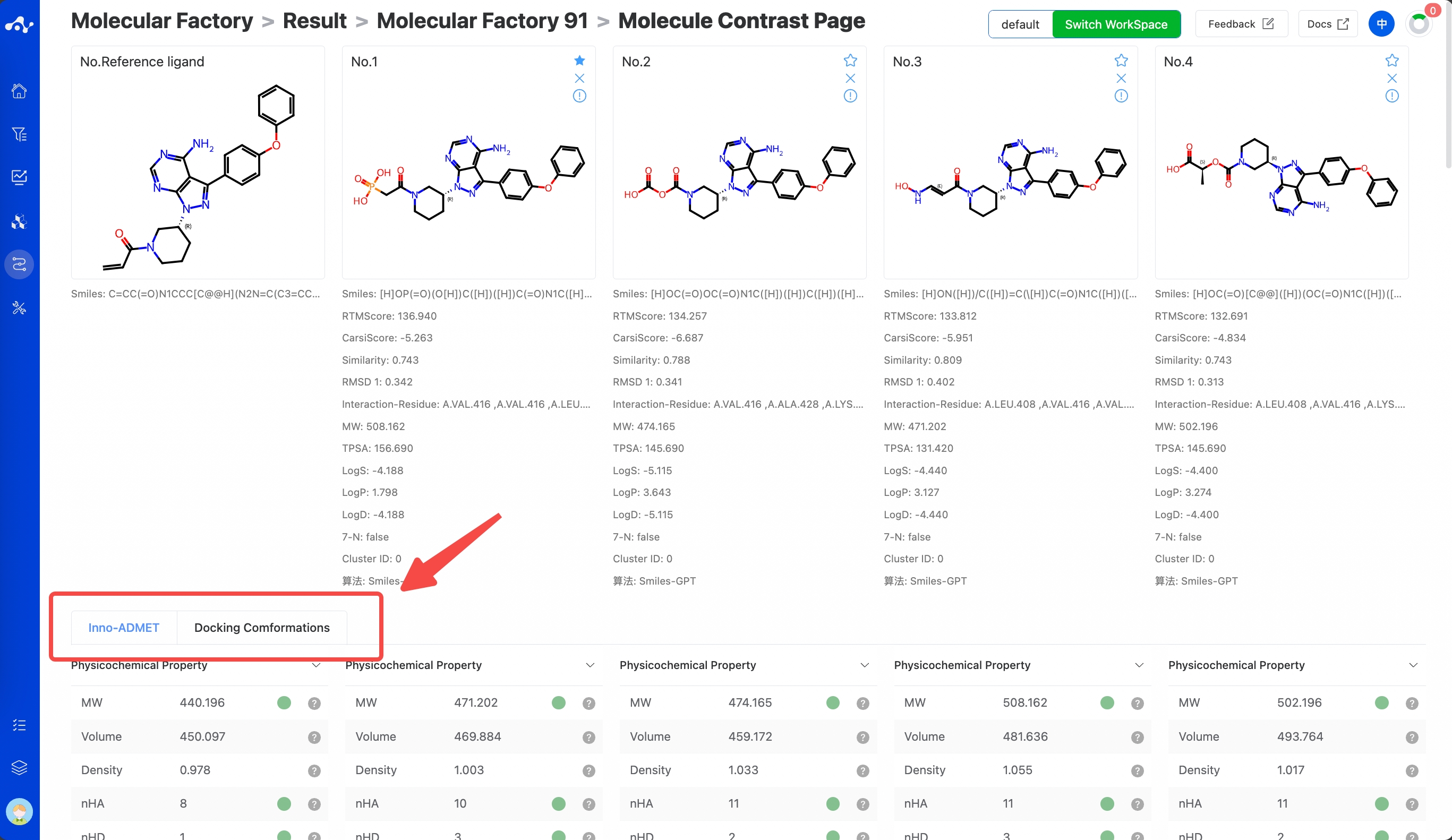
Figure 14. Molecule constrast page.
(11) Save
Click "Save", and the system will pop up a dropdown box for you to choose the file format to save (currently only supports .csv/.sdf). Once you have determined the style of the file to save, save the corresponding data to the data center as a sdf or csv file. The saved content is the molecules of the effective number displayed on the page, which are usually obtained according to your show/hide column conditions, advanced filtering conditions, favorites, or dislikes.
(12) Download
Click "Download", and the system will pop up a dropdown box for you to choose the file format to download (currently only supports .csv/.sdf). After determining the style of the file to download, the system will download the corresponding data to your local device as a sdf or csv file. The content downloaded is consistent with the save method, which also downloads the molecules of the effective number displayed on the page, which are usually obtained based on your show/hide column conditions, advanced filtering conditions, favorites, or dislikes.
(13) Create New Task
The prerequisite for creating a new task is to first save the data into a file. Before the save operation is performed, this button is disabled. As soon as the new file is saved based on the results, this button is enabled. When you click this button, the system will pop up a dropdown box for you to select the module to be calculated. After clicking, the page will immediately open a new tab and will take your saved dataset with it. After adjusting the parameters, you can submit a new task.
4. Related Literature
[1] ResGen is a pocket-aware 3D molecular generation model based on parallel multiscale modelling. Zhang, O., et al. Nature Mach Intell (2023)