Inno-Docking
1. Inno-Docking Overview
Docking, as a conventional technique in structure-based drug design, has been widely employed to identify potential hits from compound libraries. It helps us understand the binding mode between proteins and ligands and estimate their binding affinities. Typically, the reliability of a docking program mainly depends on the efficiency of its conformational search algorithm and the quality of its scoring function. The former is dedicated to generating ligand conformations, while the latter calculates the binding affinity of protein-ligand complexes.
An increasing number of studies have found that the binding affinities predicted by scoring functions embedded in docking software do not show satisfactory correlation with experimentally determined affinities and may even fail to effectively distinguish active from inactive compounds. With the rapid advancement of computer technology, scoring functions based on machine learning algorithms have been developed. These can implicitly learn and capture nonlinear protein-ligand binding features. Results have shown that, whether in terms of scoring power (ranking binding affinities), docking power (distinguishing native from decoy binding poses), or screening power (discriminating active compounds from decoys), scoring functions based on machine learning algorithms have better flexibility and higher performance than traditional scoring functions.
Therefore, the Inno-Docking module integrates not only the classic physical docking program AutoDock Vina, but also proprietary AI-based docking programs such as CarsiDock and CarsiDock-Cov. Compared to physical methods, AI docking programs have significant advantages in docking pose accuracy. Additionally, Inno-Docking provides comprehensive protein preprocessing, ligand preprocessing, intelligent automated docking parameter setup, and detailed data analysis capabilities on the results page.
2. User Guide
On the docking task creation page, the interface is divided into two areas according to function: a 3D visualization editing area and a docking parameter settings area. In the 3D visualization editing area, you can change the display style of the structure according to your habits. Hold the left mouse button to rotate the protein, hold the right mouse button to pan, and scroll the mouse wheel to zoom in and out. According to the docking workflow, the process is divided into three main steps: protein preprocessing, ligand preprocessing, and docking parameter setting.
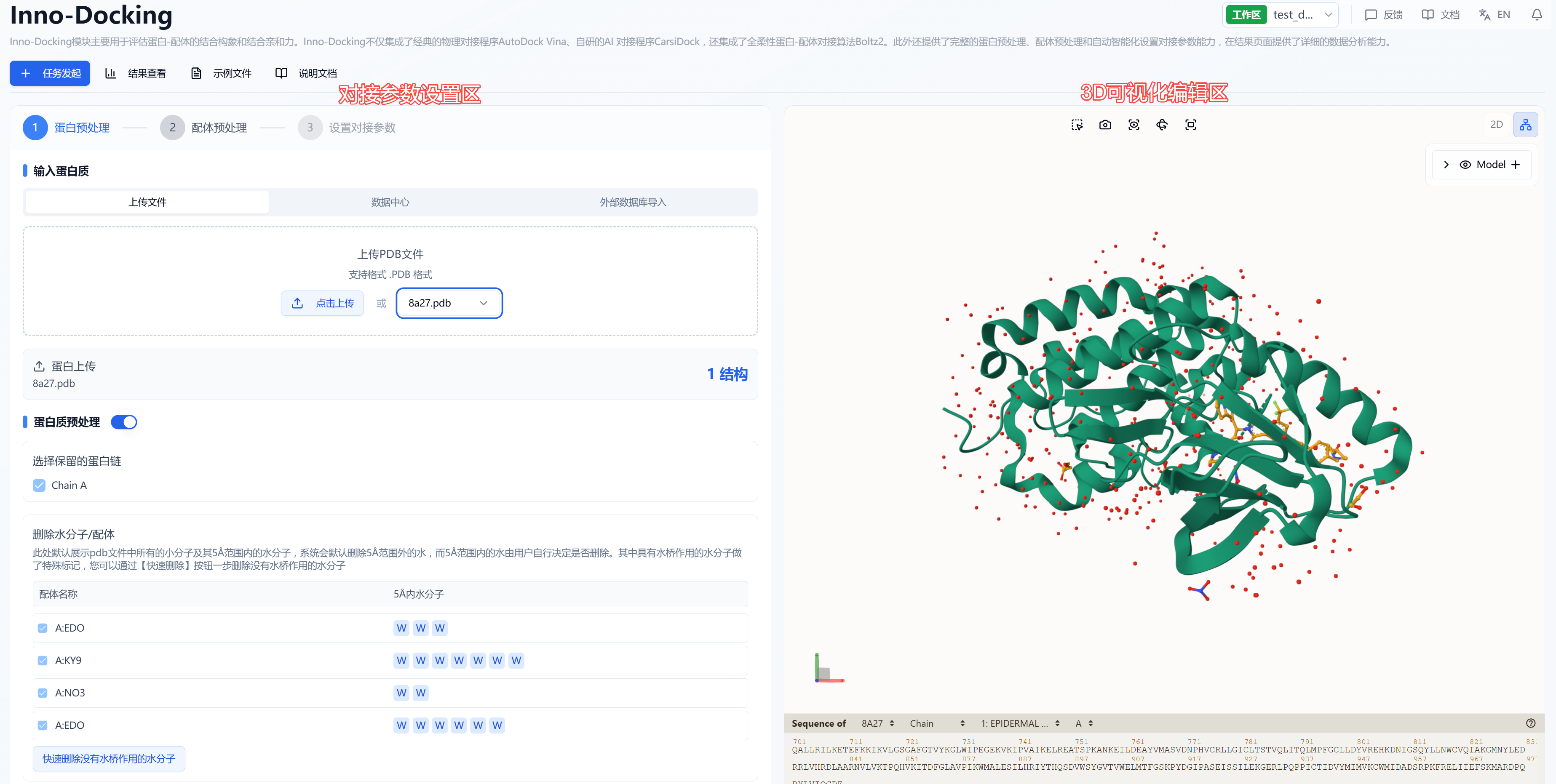
Figure 1. Docking Calculation Page Introduction
(1) Protein Preprocessing
According to conventional protein preprocessing procedures, a comprehensive set of steps is provided, including selecting retained protein chains, repairing incorrect structures/adding missing residues, adding hydrogens, and energy minimization. The settings panel is shown in Figure 2. The default selections on the page are the currently optimal parameters; users can also choose suitable parameters based on their own expertise.
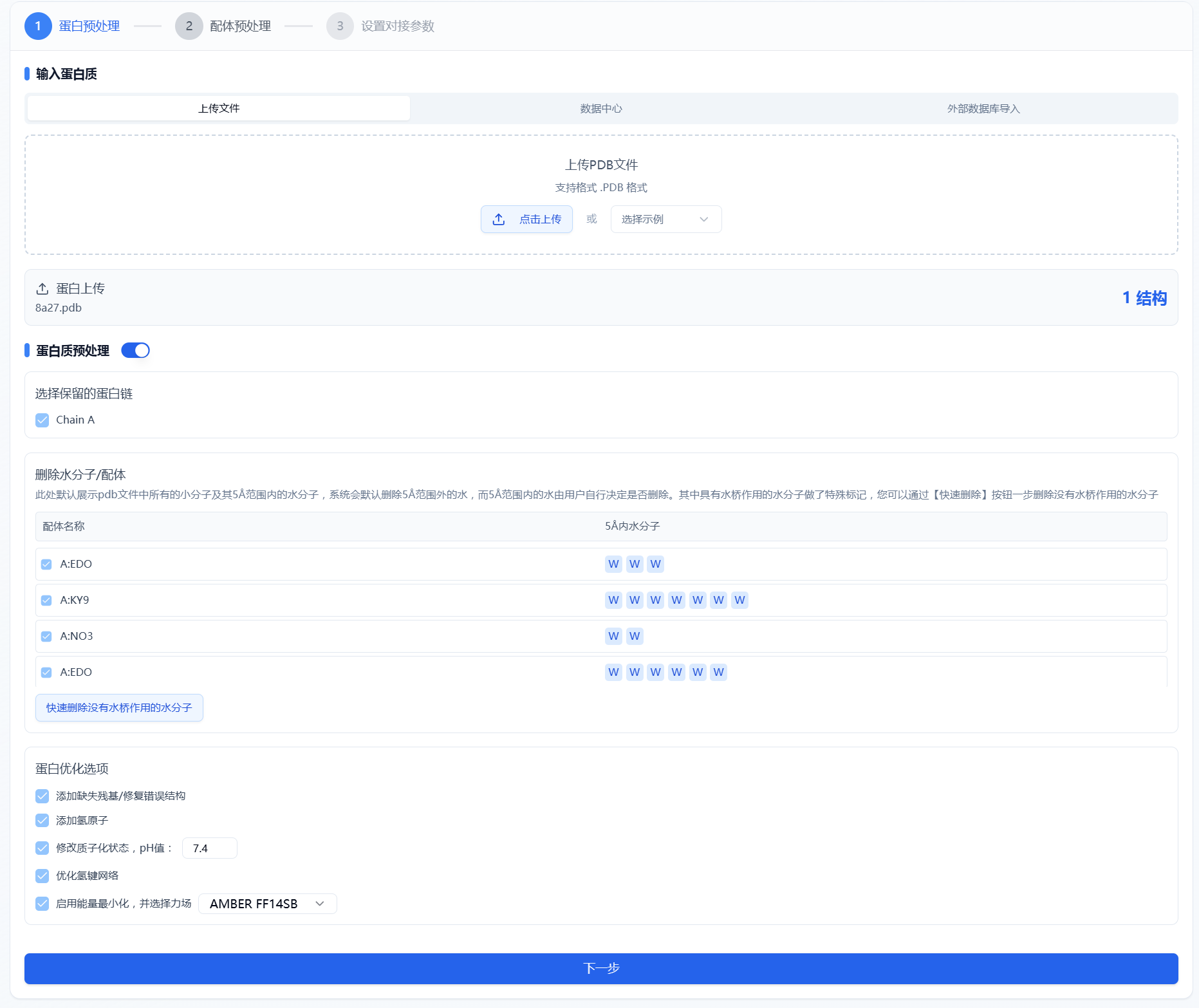
Figure 2. Protein Preprocessing
- Protein Input
The platform provides three methods to upload protein files: file upload, data center, and external database import.
File Upload: Select "File Upload" checkbox and click the upload button to select a local file. Only .pdb format is supported.
Data Center: Select "Data Center" checkbox and click below to choose a protein from the data center. Click the file name to select it, and the pop-up will disappear once done.
External Database Import: Select "External Database Import" checkbox. If the user knows the protein's PDB ID, it can be directly entered in the text box (4 digits) to download and display the structure.
Note! Currently, CarsiDock supports water molecule-based docking but does not support docking with other cofactors.
- Protein Preprocessing
Whether or not you need protein preprocessing depends on the type of protein you upload.
If the uploaded protein has already been preprocessed, you may turn off the protein preprocessing toggle and proceed to the next step.
Otherwise, it is recommended to enable the toggle to perform preprocessing operations.
Select retained protein chains: By default, all chains in the uploaded .pdb file are listed and selected. If a chain is unchecked, it will not participate in the final calculation.
Remove water/ligands: All small molecules and water molecules within 5Å are listed by default. The system will automatically remove water molecules outside 5Å, while those within 5Å are up to the user to decide. Water molecules with water bridge functions are specially marked; you can use the "Quickly remove water molecules without water bridges" button to remove them in one go. To remove a specific small molecule, uncheck it.
- Protein Optimization Options
Add missing residues/repair incorrect structures: optional, checked by default.
Add hydrogens: required.
Adjust protonation states: optional, checked by default, pH set to 7.4.
Optimize hydrogen bond network: optional, checked by default.
Enable energy minimization: optional, checked by default, with AMBER FF14SB force field selected.
- AMBER FF14SB (recommended): The FF14SB parameter set in the AMBER suite is designed for protein atom interactions and provides high accuracy and reliability, particularly for amino acid side chains and key interactions in protein folding.
- AMBER FF15IPQ: An improved parameter set compared to FF14IPQ, including more polarization effects and hydrogen bond parameters, leading to better description of protein electronic structures.
- AMBER96: An earlier version, still widely used in biomolecular simulations.
- AMBER99SB: An improved version of AMBER99 with more refined side chain parameters.
- CHARMM36: A widely used force field in biomolecular simulation, known for its accuracy in capturing protein conformations and dynamics.
(2) Ligand Preprocessing
The platform provides standard ligand preprocessing steps, including removing salts, retaining the largest molecular fragment, generating isomers (ionization states, tautomers, stereoisomers), adding hydrogens, and energy minimization. The settings panel is shown in Figure 3. The default selections are currently optimal parameters; users may adjust as needed. The settings are consistent with those in the "Ligand Preprocessing" module.
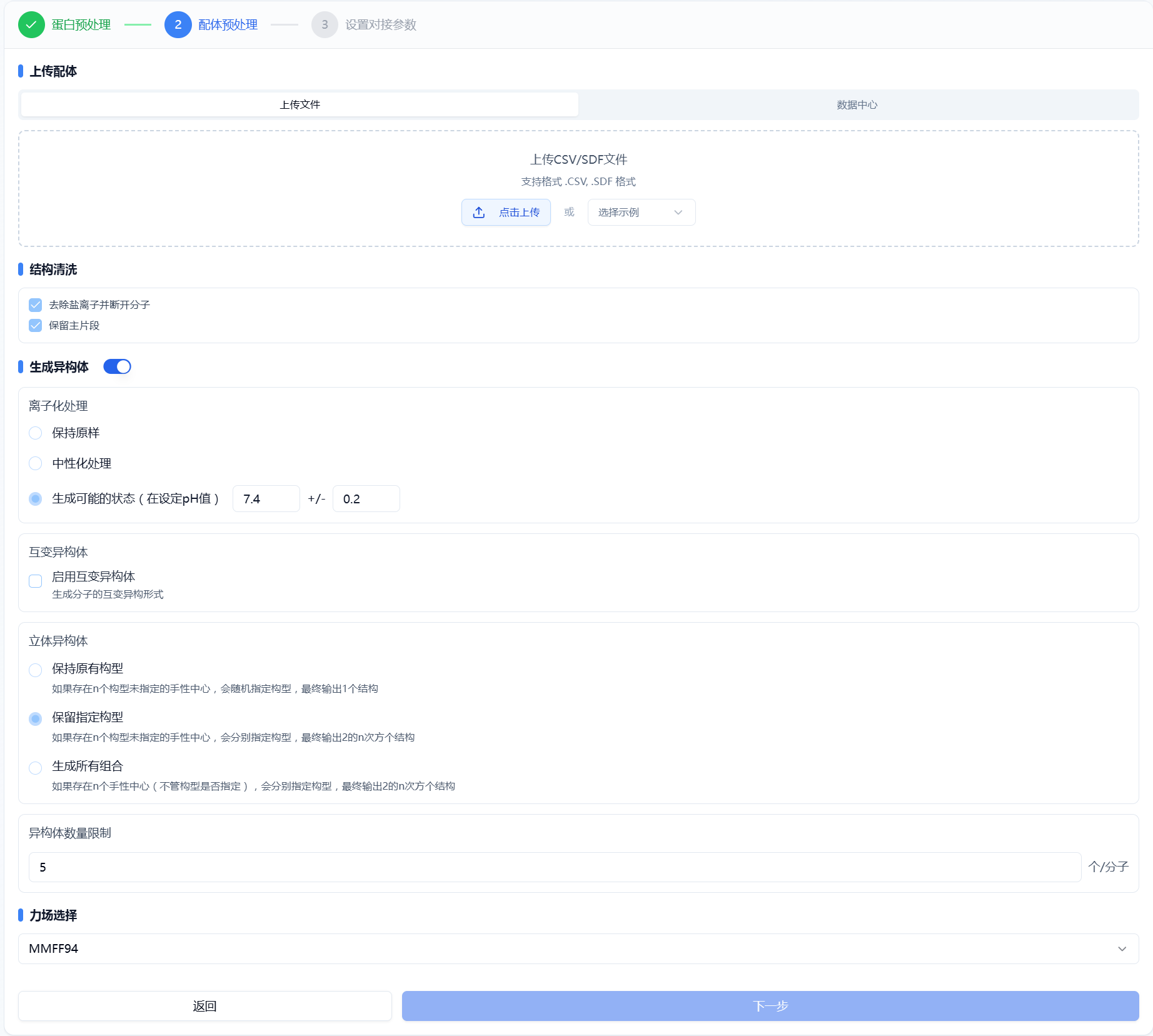
Figure 3. Ligand Preprocessing
- Upload Ligand
Currently, only the following methods are supported:
- File Upload: Select its checkbox and use the button to select local files. Supported formats are .csv and .sdf.
- Data Center: Select its checkbox and use the button to select from the data center via a pop-up window.
- Structure Cleaning
Process the uploaded structure, including removing salts, disconnecting molecules, and retaining the main fragment.
- Isomer Generation
By default, the uploaded molecules are enumerated (toggle is “on”) to generate more isomers, including ionization, tautomers, and stereoisomers. When the toggle is “off”, the system keeps the original conformations with no other processing.
- Ionization: Generates likely ionization states by adjusting pH range.
- Tautomers: Generates likely tautomers based on ionization state.
- Stereoisomers: Generates likely stereoisomers based on molecular chirality.
- Isomer Limit: The default is up to 5 isomers. Users can adjust this as needed.
- Force Field Selection
- MMFF94: A professional small molecule force field, regarded as one of the most accurate.
- UFF: The Universal Force Field covers the entire periodic table—a general-purpose force field for situations where a dedicated force field is unavailable.
(3) Docking Parameter Settings
In this step, users need to first choose a docking method and then set parameters according to the selected method.
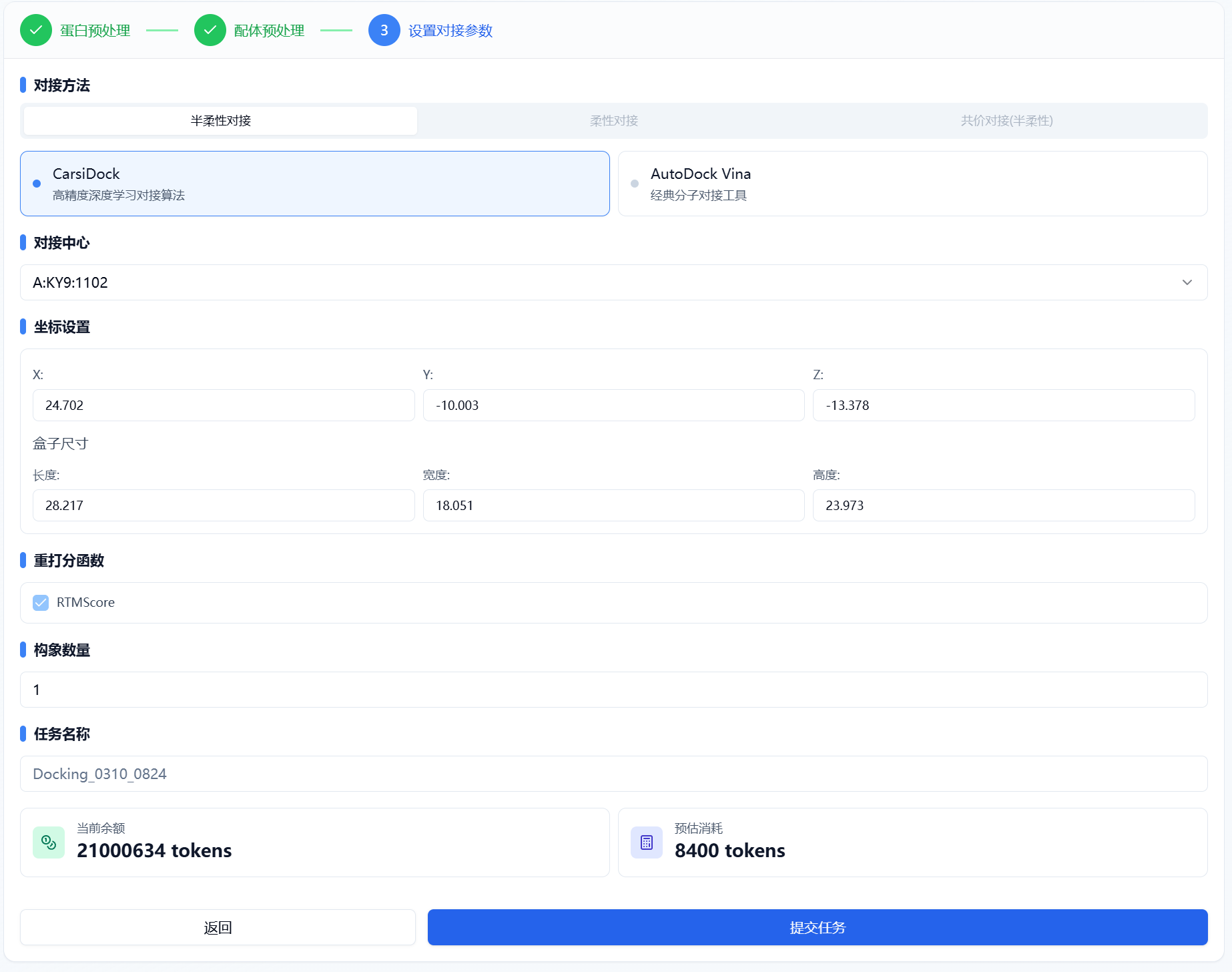
Figure 4. Docking Parameter Settings
- Docking Methods
Three docking types are currently supported: semi-flexible docking, flexible docking, and covalent docking.
- Semi-flexible Docking: The receptor conformation remains fixed, allowing ligand conformational sampling and optimization within the binding pocket. Two methods are offered: CarsiDock (a proprietary fast, high-accuracy AI algorithm) and AutoDock Vina (an open-source algorithm that has been optimized for speed and accuracy over the default version).
- Flexible Docking: Based on the Boltz-2 flexible docking tool, this considers conformational changes for both receptor and ligand during docking, leading to higher accuracy but with greater computational cost.
- Covalent Docking: Simulates covalent bond formation between ligand and receptor, based on the proprietary CarsiDock-Cov algorithm, supporting 10 common covalent reaction types. The user must specify the covalent residue and reaction type.
- Docking Sites
Both CarsiDock and AutoDock Vina support two methods of defining docking sites: selecting a ligand in the complex or manually specifying docking coordinates. The default coordinates and box size are set by the system.
- Docking Center: You may select an existing ligand as the docking center. If multiple ligands are present, the system displays the largest by mass and allows selection via the drop-down. The geometric center (XYZ) and box size (+5Å from edge atoms) are calculated automatically.
- Custom Docking Site: Users may specify custom sites by clicking a residue in the 3D viewer, which then displays its coordinates on the parameter panel. Default box sizes are provided, but users should set appropriate values based on knowledge of the pocket, as too small/large sizes affect accuracy and calculation time.
- Rescoring Function
Uses the proprietary RTMScore rescoring function by Carbon Silicon Intelligence (default: enabled).
- Number of Poses
By default, each isomer outputs one docking pose. Users can adjust this up to a maximum of 100.
(4) Monitoring Progress and Viewing Results
After submitting the task, the page automatically switches to the "Results" subpage, where you can track the task's run status (progress bar) for the current module. You can also view running tasks from all modules via the "Notifications" dropdown in the upper right. For large datasets, the system calculates in batches, so once a batch finishes (even if the overall task is still running), you can click "View Results" to access completed predictions (incomplete molecules are not shown), and refresh to get the latest results.
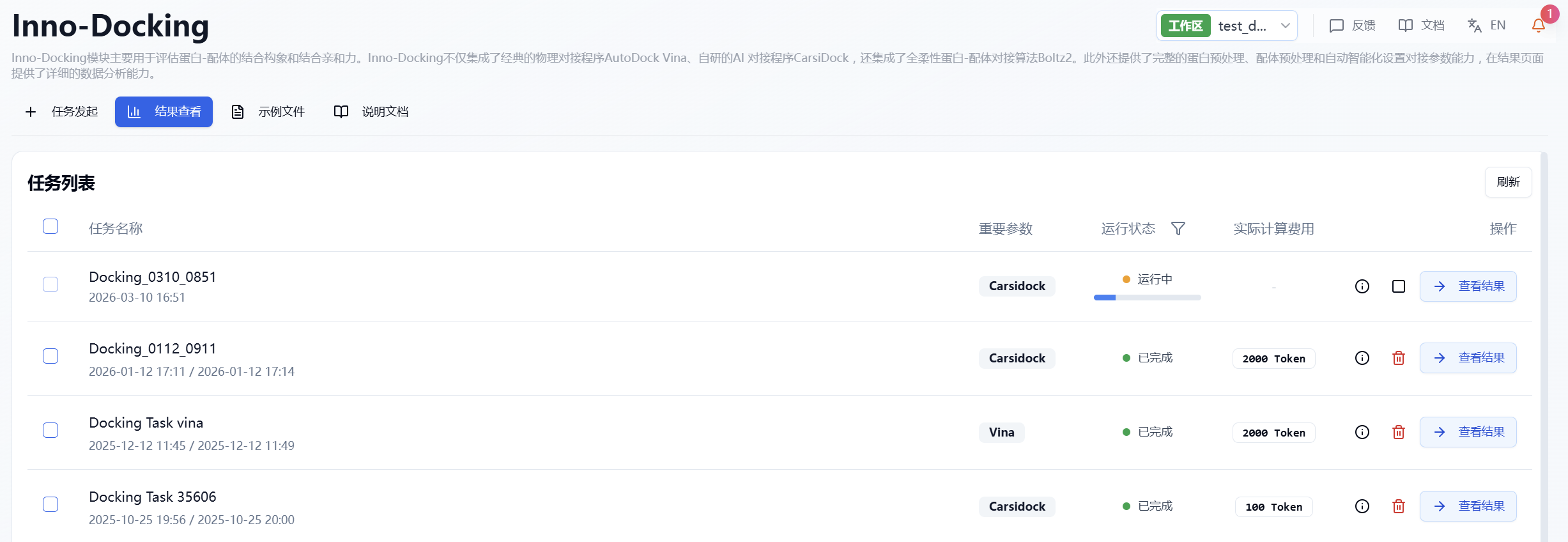
Figure 5. Viewing Results
3. Results Analysis
The results page is divided into a top summary, a molecule list on the left, and a protein visualization area on the right. By default, the detailed results area shows the list view, but you may switch to a grid view. The grid view offers a concise presentation of molecular structures, while the list view provides detailed calculated results for data analysis. The protein visualization area is fixed content and always displays the protein-ligand binding mode, regardless of the left page. Users can quickly browse molecular interactions using the "<" and ">" buttons at the bottom.
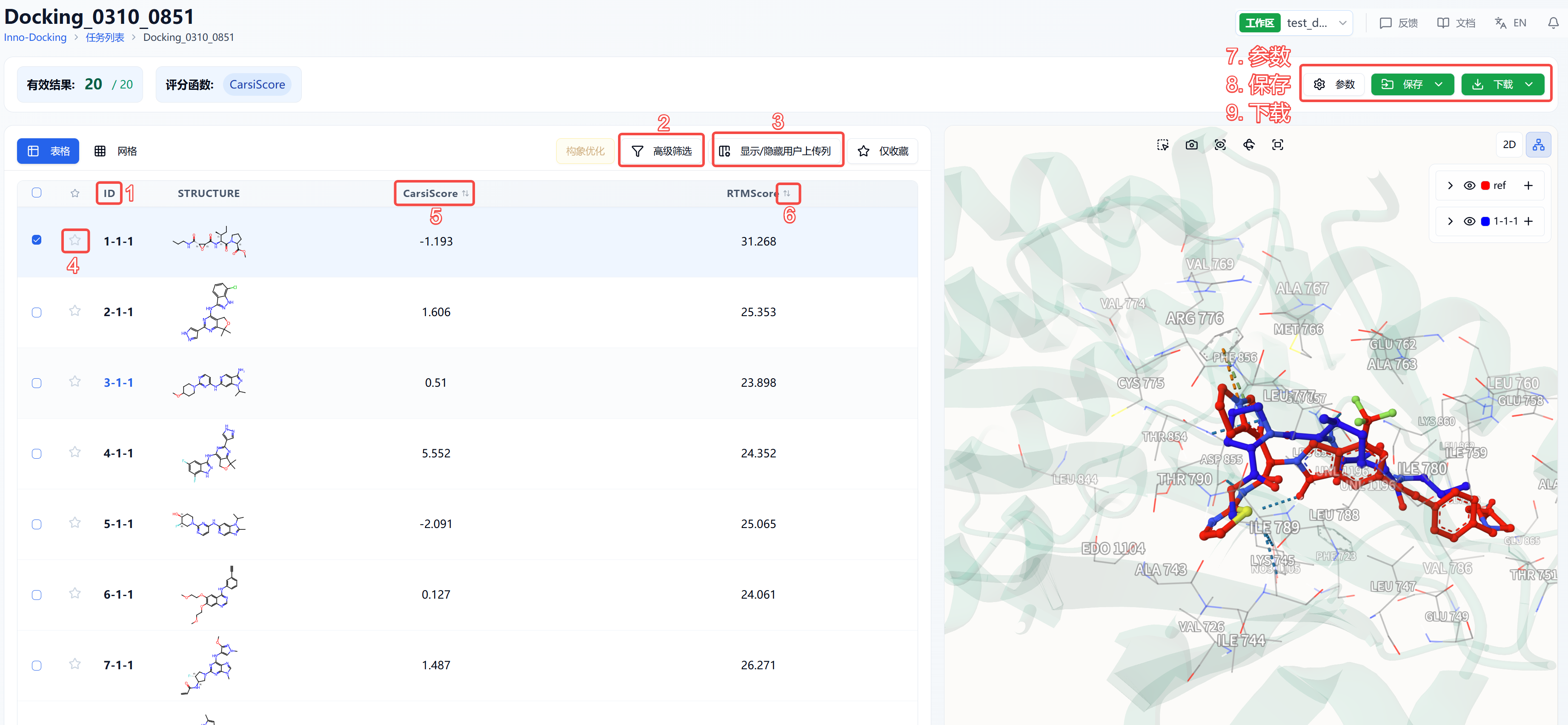
Figure 6. Results Page Functional Distribution
(1) Meaning of ID in Table
ID is assigned according to the molecular order in the original file. If no isomers are generated, IDs are consecutive from 1-N; if isomers are generated, the ID format is X-Y-Z, where X is the order of molecules, Y is the isomer number (smaller Y = higher likelihood), and Z is the pose number.
(2) Advanced Filtering
Advanced filters allow for selecting molecules whose properties fall within specified ranges, excluding molecules that do not meet expectations. After filtering, the page displays only molecules meeting the filter criteria.
(3) Show/Hide User Uploaded Columns
By default, result lists do not display uploaded file information. Toggling the show/hide uploaded columns option updates the view. At the top, "Select All" and "Deselect All" shortcuts are provided for convenience.
(4) Favorites
This feature helps users mark favorite molecules. Clicking the favorite icon lights it up, indicating it is marked. Clicking "Show only favorites" in the table will filter the list to display only those marked. Favorites can be unmarked by clicking the icon again.
(5) Property Explanations
Hovering over any property name displays its explanation.
(6) Sorting
Click property names in the results table to sort the data, e.g., RTMScore; clicking repeatedly toggles ascending, descending, and original order.
(7) Parameters
Displays details of the docking task parameters.
(8) Save
Clicking "Save" triggers a dropdown to select a file to save to the data center. Options include "Preprocessed Results" and "Post-docking Results". "Preprocessed Results" refer to protein and ligand structures after preprocessing but before docking. "Post-docking Results" refer to results output by the docking program. The saving range for post-docking results includes: selected rows (rows checked in the table), filtered rows (those passing advanced filters), and all rows (all results).
(9) Download
Clicking "Download" opens a dropdown to select a file to download locally. Options include "Preprocessed Results" and "Post-docking Results" as above. Download ranges are: selected rows, filtered rows, and all rows.
4. Algorithm Introduction
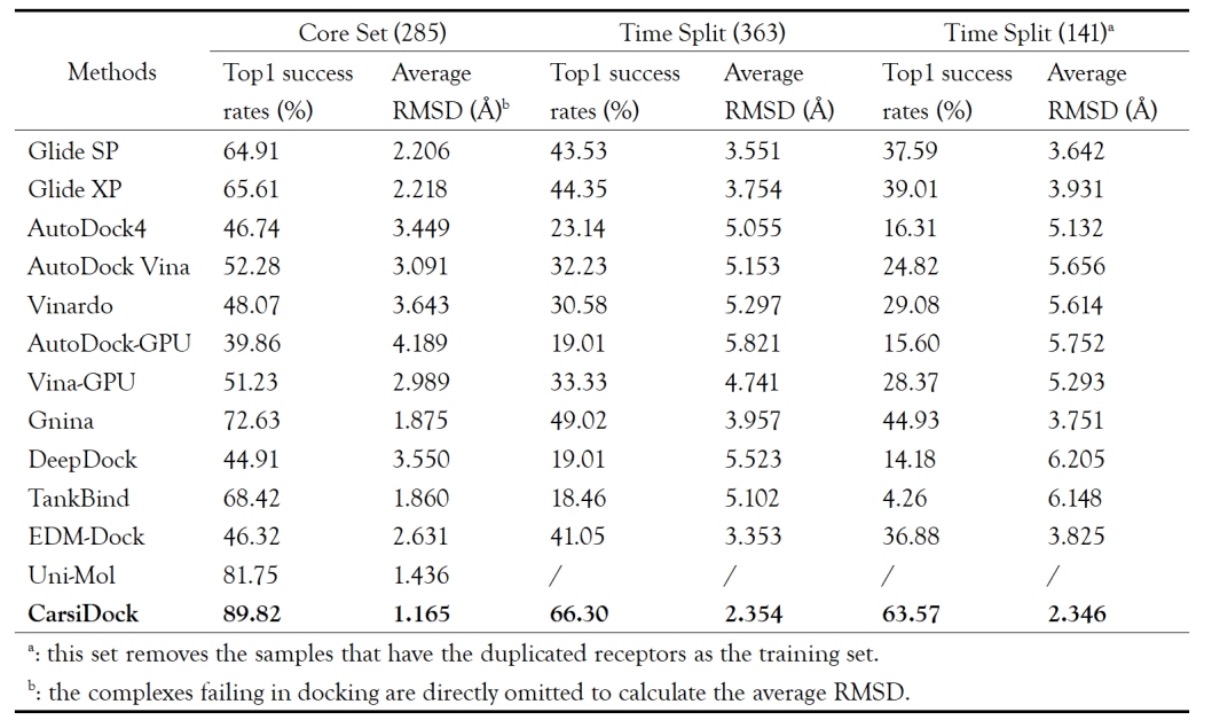
CarsiDock is a semi-flexible AI docking method developed by Carbon Silicon Intelligence. It pioneered a rigid docking-guided self-distillation approach and is pre-trained with large-scale physical simulation data. Inspired by pre-training techniques such as BERT/ChatGPT, CarsiDock effectively integrates physical docking with AI approaches: first predicting the protein-small molecule binding mode with AI, then rapidly obtaining the docking pose via gradient descent. Massive docking data are generated by physical methods, with structures, interaction patterns, and binding affinities, providing a robust pre-training foundation for AI docking. Crystal data is used for further fine-tuning to enhance predictive accuracy. Results show that CarsiDock ensures topological reliability of binding poses and can reproduce key interactions seen in crystal structures, making it broadly applicable with high docking and screening performance.
CarsiDock-Cov is an automated covalent docking method based on deep learning. Building upon the original non-covalent CarsiDock, it integrates modules for covalent bond constraints, geometry optimization, reactive group identification, and ligand covalent modification, thereby enabling efficient, automated docking of covalently bound ligands. Validation on multiple public datasets demonstrates that CarsiDock-Cov outperforms many existing covalent docking tools in pose prediction and virtual screening, especially showing better generalization in complex cross-docking scenarios.
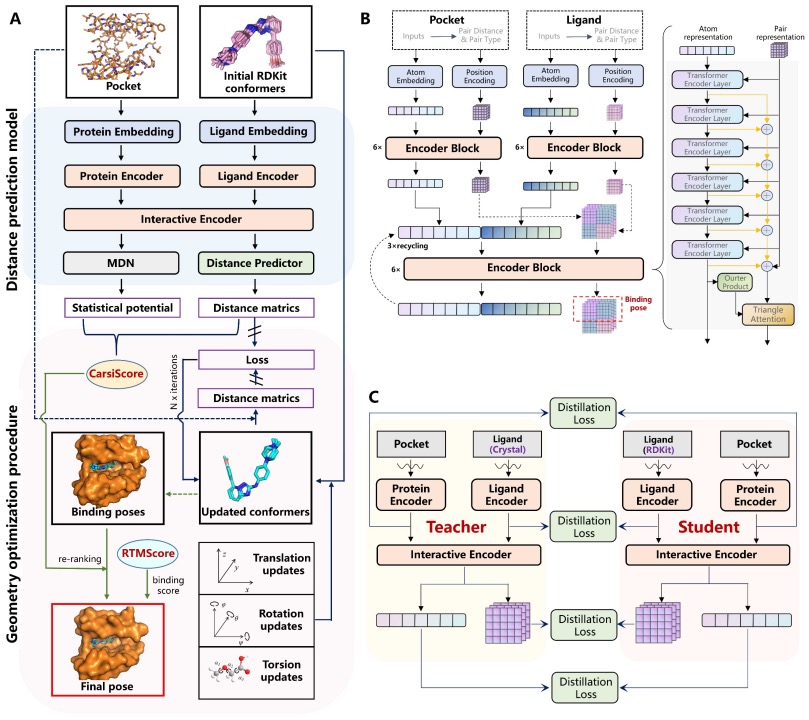
Figure 7. CarsiDock Model Framework
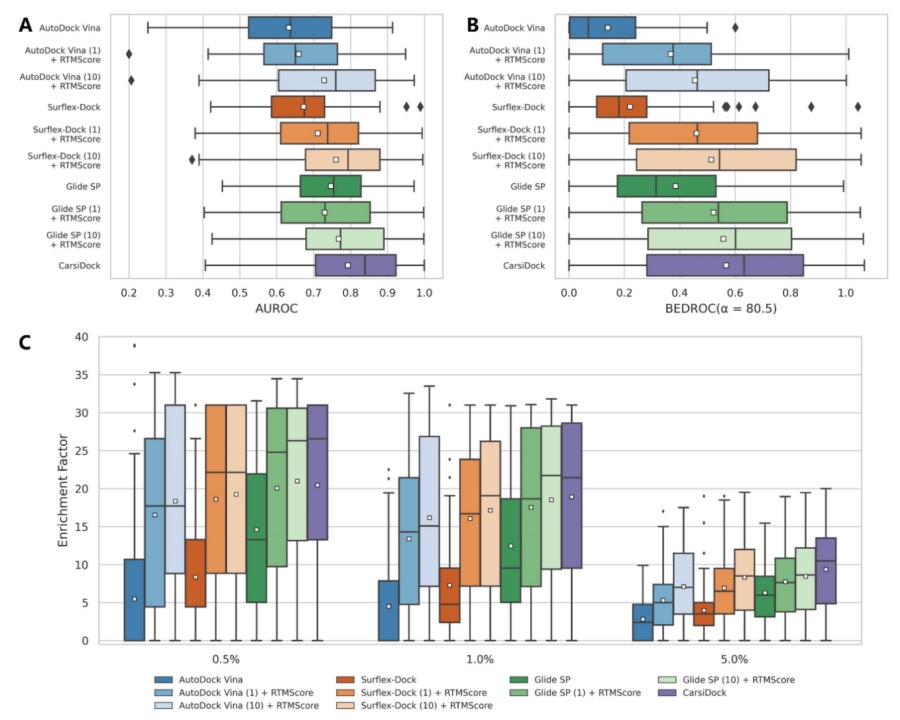
Figure 8. Screening Performance of Multiple Methods on the DEKOIS2.0 Dataset
Table 1. Comparison of Top1 Success Rate and Mean RMSD for PDBBind CoreSet and Fractional Datasets
5. References
[1] CarsiDock: a deep learning paradigm for accurate protein-ligand docking and screening based on large-scale pre-training, Chemical Science, 2024, 15, 1449-1471.
[2] CarsiDock-Cov: A deep learning-guided approach for automated covalent docking and screening, Acta Pharmaceutica Sinica B, 2025, 15, 5758-5771.