Inno-Rescoring
1. Inno-Rescoring 概述
Inno-Rescoring 模块提供了基于机器学习算法的优秀打分函数。为了方便用户可以更直接的使用我们的重打分函数,Rescoring 模块支持用户在本地对接后上传相关的文件进行重打分,用户可以选择重打分函数重新评估蛋白质-配体结合姿势的结合亲和力。
2. 使用说明
用户只需要四个步骤就可以完成计算:上传对接后的蛋白文件-上传对接后的配体文件-任务名称(可忽略)-提交任务(必点)。
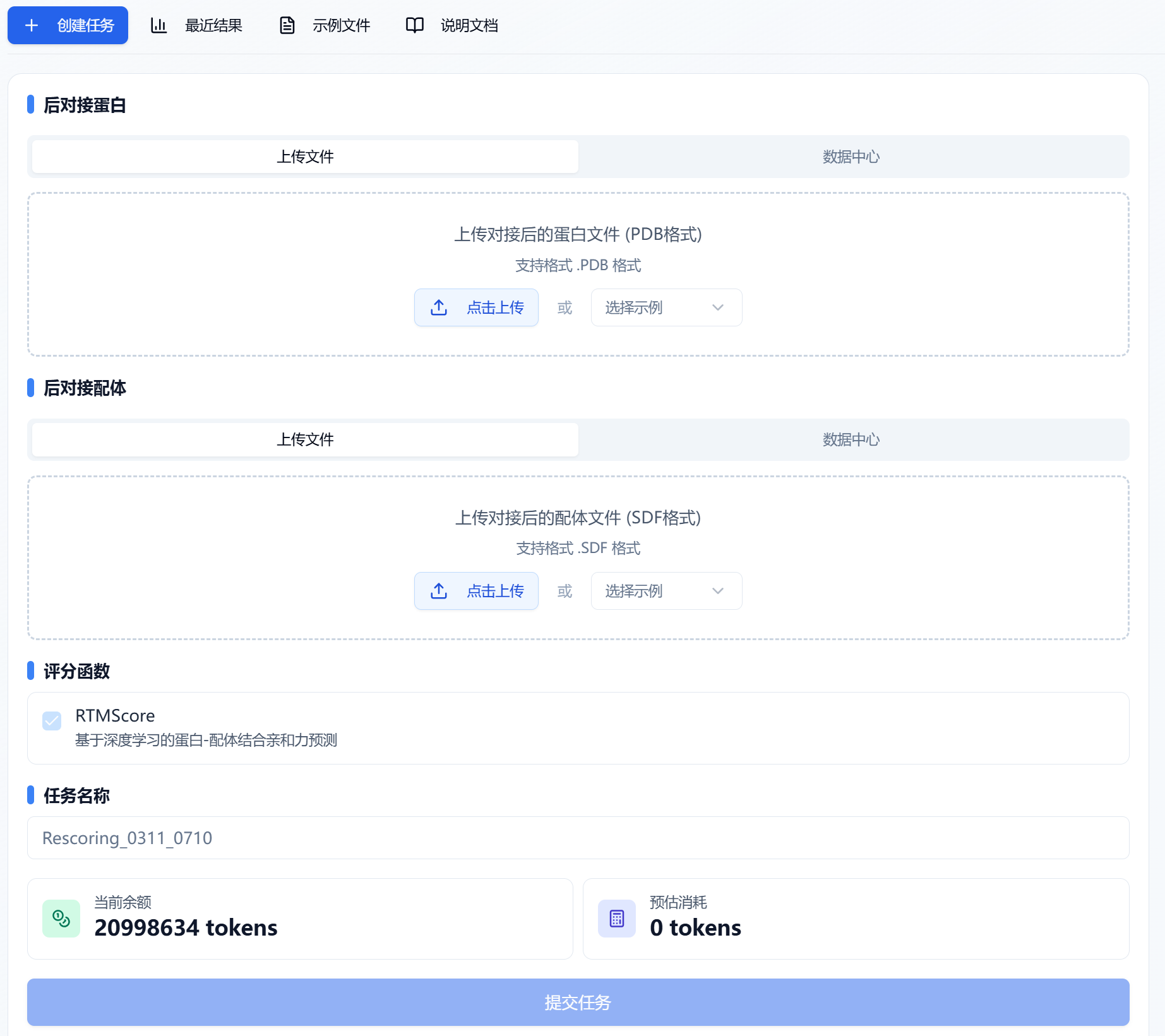
图 1. 上传后对接蛋白和后对接配体——上传文件/数据中心
(1) 上传后对接蛋白
平台提供了两种上传蛋白的方式:上传文件和数据中心。
- 上传文件
复选框选中“上传文件”,通过点击下方按钮选择本地文件即可。
- 当前支持的文件格式: .pdb
- 数据中心
复选框选中“数据中心”,通过点击下方按钮页面出现弹窗,点击文件名称来选择数据中心的数据,点击完之后弹窗消失。
(2) 上传后对接配体
平台提供了两种上传对接后配体的方式:上传文件和数据中心。
- 上传文件
复选框选中“上传文件”,通过点击下方按钮选择本地文件即可。
- 当前支持的文件格式: .sdf
- 数据中心
复选框选中“数据中心”,通过点击下方按钮页面出现弹窗,点击文件名称来选择数据中心的数据,点击完之后弹窗消失。
在确定好所有的参数后,命名任务名称,点击提交即完成任务提交操作。
(3)运行进度和结果查看
提交任务后,页面会自动跳入当前页面的“最近结果”子页面中,您可以在该页面查看当前模块的任务运行状态(进度条),也可在右上角的“通知”下拉框中查看所有模块正在运行的任务。当数据量较大时,系统会分批计算,因此只要有一批数据算完后(整个任务还在运行中),即可点击“查看结果”按钮进入结果页面,查看当前已完成计算的预测结果列表(未完成计算的分子暂不显示),并且可以在当前页面通过刷新来获取最新算完的数据。
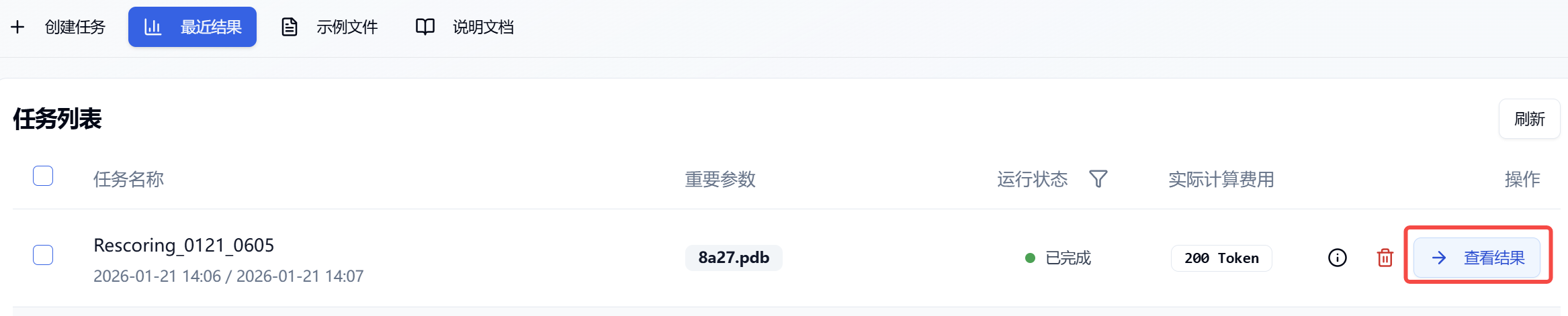
图 2. 查看结果
3. 结果分析
结果页面默认状态下是以表格显示,可点击网格切换显示形式。您可以查看所有得分,并对这些得分进行排序。
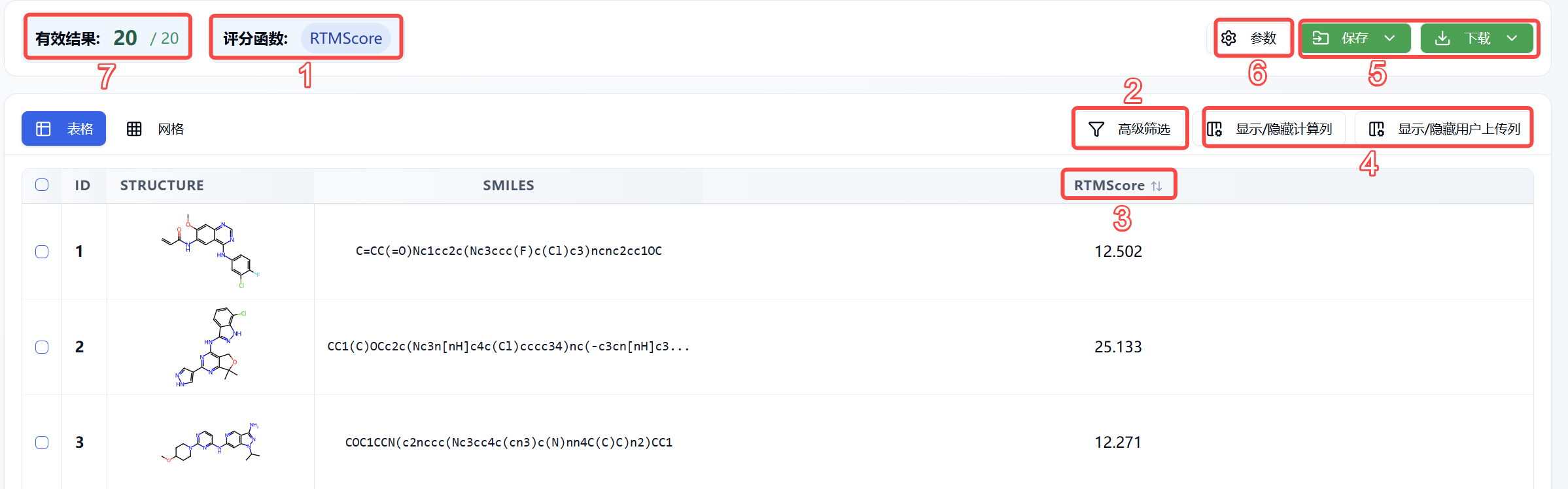
图 3. 结果页面功能分布
(1) 评分函数
评分函数为 RTMScore。
(2) 高级筛选
高级筛选提供了范围筛选,可进一步筛选出得分值指定范围内的分子,以排除不符合预期结果的分子。
(3) 重打分函数解释/排序
把鼠标移入重打分函数的名称上,可查看该重打分函数对应的解释;点击重打分函数名字可重新排序,如 RTMScore,点击一次为升序,再点一次为降序,再点一次即恢复原始排序。
(4) 显示/隐藏列
可显示/隐藏计算列和用户上传列。
(5) 保存/下载
点击保存/下载,系统将弹出下拉框让您选择保存/下载的文件格式(目前仅支持.csv/.sdf)。确定好保存/下载的文件格式后,可设置保存范围,包括勾选的行--即表格首列的方框勾选分子;筛选后的行--即通过高级筛选得到的分子;所有行--即所有的分子;当前显示的列--即当前显示在页面上的列;所有列--即包括显示和未显示的所有列。系统将根据您的设置,保存相应的数据至数据中心,或下载数据至本地。
(6) 参数
显示该任务计算时所使用的参数。
(7) 有效结果
显示格式为 当前页面显示分子(此处即通过高级筛选后得到的分子数)/总分子。
4. 相关算法介绍
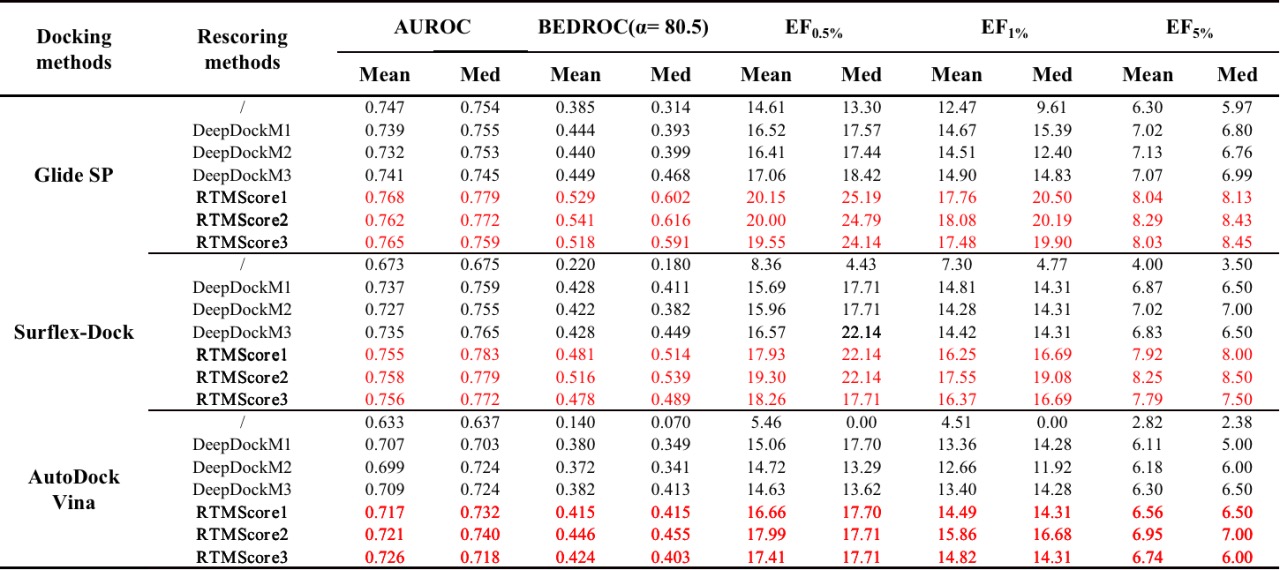
RTMScore 是一种新型的蛋白质-配体结合亲和度预测评分函数,我们通过 Graph Transformer 提取蛋白质的氨基酸残基以及配体的原子节点特征,并通过混合密度网络(Mixture Density Network, MDN)获取蛋白质各氨基酸残基和配体各原子间距离的概率密度分布,最后将其转化为统计势以用于蛋白-配体间结合强度的评估。结果表明,RTMScore 的对接能力和筛选能力在 CASF-2016 标准数据集上显著超越了当前的其他主流方法,其在有无天然构象的存在下分别可取得 97.3%和 93.4%的平均 top1 对接成功率(DeepDock 和 PIGNet 在无天然构象时仅为 87.0%);而在筛选能力评估中可取得 66.7%的平均 top1 成功率和 28.00 的 1%富集因子(DeepDock 和 PIGNet 的两项指标分别为 55.4%、19.60 以及 43.9%、16.41)。同时我们进一步在 DEKOIS2.0 数据集上评估了 RTMScore 筛选能力,结果表明 RTMScore 的富集能力明显优于以相似策略构建所得的 DeepDock 和经典方法 Glide SP,详见参考文献[1]。
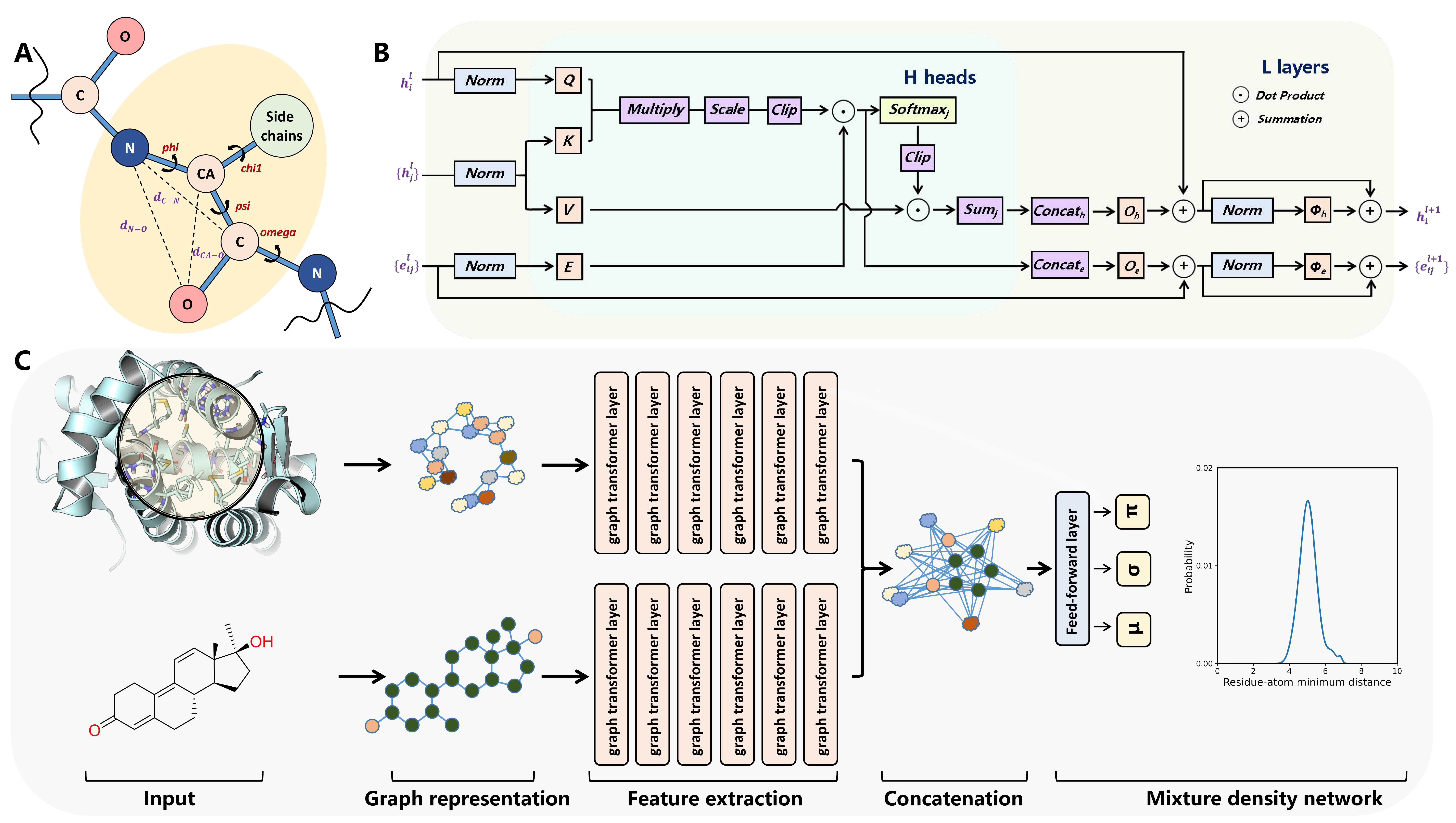
图 4. RTMScore 模型框架
表 1. RTMScore 与其他先进方法在 CASF-2016 基准测试上的对接能力和筛选能力的比较
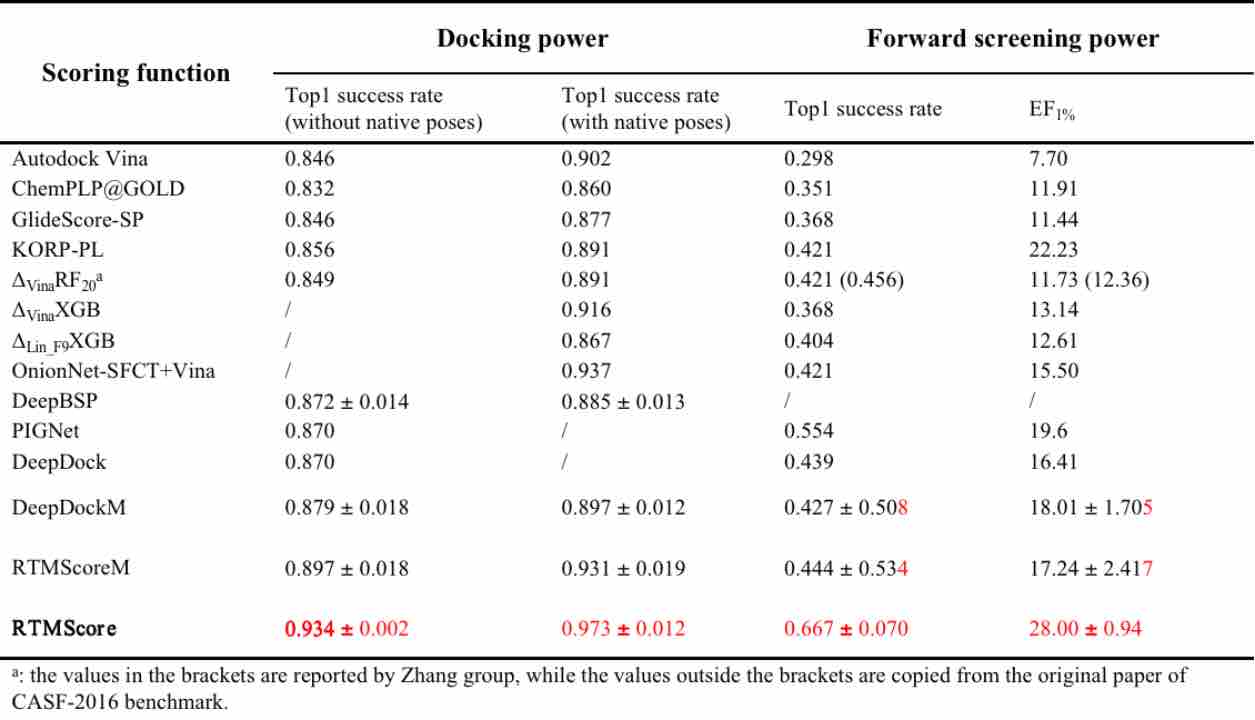
表 2. RTMScore 在 DEKOIS2.0 数据集上的筛选能力评估结果
5. 相关文献
[1] Boosting Protein-Ligand Binding Pose Prediction and Virtual Screening Based on Residue−Atom Distance Likelihood Potential and Graph Transformer, Journal of Medicinal Chemistry, 2022, 65, 10691-10706.