Inno-PepDocking
1. Inno-PepDocking 概述
Inno-PepDocking 是基于 RAPiDock 的多肽对接的模块,这是一种基于扩散生成模型的蛋白质-多肽对接方法,能够在全原子水平上实现快速、准确且合理的分子对接。在 RefPepDB-RecentSet 测试集上达到了 93.7%的 Top-25 预测成功率,较 AlphaFold2-Multimer 提升 13.4%,且预测速度提升约 270 倍(每个复合物仅耗时约 0.35 秒)。支持对包括多种翻译后修饰在内的 92 种氨基酸残基进行全原子建模。
2. 使用说明
(1)蛋白预处理
输入蛋白质
- 上传文件:从本地上传.pdb 文件。
- 数据中心:从数据中心选择.pdb 文件。
- 外部数据库导入:输入 PDB ID,从 PDB 数据库中下载对应的文件。
蛋白质预处理
根据您上传的蛋白类型来确定是否进行蛋白预处理。 如果上传的蛋白已经做过蛋白预处理,您可以直接点击下一步。 如果没有,建议您打开开关进行预处理相关的操作。
选择保留的蛋白链:此处默认显示上传的.pdb 文件中所有链的信息,且所有链都被勾选。当某条链被取消勾选后,将不再参与后续的计算。
删除水分子/配体:此处默认展示 .pdb 文件中所有的小分子及其 5Å 范围内的水分子。系统会默认删除 5Å 范围外的水,而 5Å 范围内的水由用户自行决定是否删除。其中具有水桥作用的水分子做了特殊标记,您可以通过【快速删除没有水桥作用的水分子】按钮一步删除没有水桥作用的水分子。取消勾选的配体及后边的水分子,将不再参与后续的计算。
蛋白优化选项 添加缺失残基/修复错误结构:可选项,默认为勾选状态; 添加氢原子:可选项,默认为勾选状态; 调整质子化状态:可选项,默认为勾选状态,且 pH 为 7.4; 优化氢键网络:可选项,默认为勾选状态; 能量最小化:可选项,默认为勾选状态,且选择的为 AMBER FF14SB 力场;
AMBER FF14SB(推荐使用):FF14SB 是 AMBER 程序包中的一个蛋白质力场参数集,用于描述生物分子中的原子相互作用。它是 AMBER14 程序包中的一种特别适用于蛋白质体系的力场参数集,包括描述蛋白质中氨基酸侧链和蛋白质折叠中重要残基之间相互作用的额外参数。AMBER FF14SB 在描述蛋白质的构象和动力学性质方面具有较高的精度和可靠性。
AMBER FF15IPQ:FF15IPQ 是 AMBER 程序包中的一种改进的蛋白质力场参数集,相比于 AMBER FF14IPQ 具有更高的精度和可靠性。AMBER FF15IPQ 包括更多的偏极化效应和氢键参数,可以更准确地描述蛋白质的电子结构。
AMBER96:AMBER96 是 AMBER 程序包中的早期版本,经过多年的发展和优化,现在已有更新的版本,如 AMBER14 和 AMBER18 等。但是,AMBER96 仍然被广泛应用于生物分子模拟领域,特别是对于早期的研究和一些经典案例的模拟。
AMBER99SB:AMBER99SB 是 AMBER99 力场的一个改进版本,包括了描述蛋白质中氨基酸侧链和蛋白质折叠中重要残基之间相互作用的额外参数。
CHARMM36:CHARMM36 在描述蛋白质的构象和动力学性质方面具有较高的精度和可靠性。它在蛋白质和蛋白质-配体相互作用等领域的研究中被广泛应用,是生物分子模拟领域中常用的力场参数集之一。
(2)多肽输入
- 上传文件:从本地上传.fasta, .fas, .fa, .txt 格式文件。
- 数据中心:从数据中心导入 fasta。
- 文本输入:直接输入多肽序列文本,格式:>序列名称\n 序列内容,支持多个序列。 多肽输入支持的氨基酸类型包括 20 种标准氨基酸和 72 种非标准氨基酸,如下所示: • 20 种标准氨基酸:G, A, V, L, I, P, F, Y, W, S, T, C, M, N, Q, D, E, K, R, H • 72 种非标准氨基酸,使用[XXX]格式,如:RMF[HYP]R[PTR]NAPYL HYP, SEP, TYS, ALY, TPO, PTR, DAL, MLE, M3L, DLE, DLY, AIB, MSE, DPR, MVA, NLE, MLY, SAR, ABA, FME, DAR, ORN, CGU, DPN, DTY, DTR, 4BF, DGL, DCY, MK8, MP8, GHP, ALC, BMT, MLZ, DVA, 3FG, DAS, 7ID, DSN, AR7, MEA, PHI, MAA, LPD, KCR, PCA, DGN, 2MR, DHI, ASA, MLU, YCP, DSG, DTH, OMY, FP9, DPP, HCS, SET, DBB, BTK, DAM, IIL, 3MY, SLL, PFF, HRG, DIL, DNE, MED, D0C
(3)设置对接参数
对接方法
选择对接方法,目前只有一种对接方法,即 RAPiDock。
对接中心&坐标设置
支持两种方式定义对接中心,分别为选择复合物中的多肽配体和自定义对接位点。默认看到的坐标是系统随机分配,长宽高为系统默认值,这几个参数代表了结合口袋的位置和大小。
- 选择复合物中的多肽配体为对接位点。 该选项只适用于复合物中自带多肽配体的情况。当复合物中有多个多肽配体的时候,系统默认显示分子质量最大的,用户可以在下拉框中切换其他多肽配体。系统会根据多肽配体的大小计算出其几何中心(XYZ 坐标),并设置合适的盒子尺寸。
- 自定义对接位点。 在自定义模式下,用户可直接把鼠标放入 3D 显示区,当您单击某残基后,参数面板上才会显示出该残基相应的坐标信息,同时系统也给了一个默认的长宽高。但是,口袋具体的大小需要用户在了解蛋白口袋的情况下自行设置,口袋太小会导致计算结果不准确,口袋太大将会增加计算的时间。
构象数量
设置对接后,每个多肽配体最多输出的构象数量。
任务名称
可自定义或者使用系统分配的默认任务名。
3. 结果分析
计算结果页面如图 1 所示。 左上方
- 有效结果:当前表格中显示数量/总共输出的数量。
- 结合自由能:即 MM-PBSA。MM-PBSA 是分子模拟领域中一种非常流行的方法,用于计算结合自由能。它被广泛应用于研究蛋白质与配体(药物分子)、蛋白质与蛋白质、蛋白质与核酸等生物分子之间的相互作用强度。该值越低,代表结合越稳定。
- 序列图例:即下方表格 SEQUENCE 列的氨基酸字符颜色,分为疏水性、极性、正电荷、负电荷、甘氨酸五类。
- 自对接评估:当设置对接参数时,如果选择以复合物中的多肽配体为中心,会对该配体进行自对接,并会显示自对接评估及在下方显示自对接结果(如果表格里输出多个自对接结果,那么自对接评估里的数值对应的是 MM-PBSA 分数最低,即结合最稳定的结构)。
左下方
- ID:即多肽输出的 ID,第一个是不同多肽序列的编号,第二个是同一序列不同构象的编号。
- SEQUENCE:输入多肽的序列。
- 结合自由能:即 MM-PBSA,点击可根据此列进行排名,点击第三次恢复初始的排序。
- 高级筛选:可根据 结合自由能 的数值对下方表格里的结果进行筛选。
- 仅收藏:筛选表格中收藏的结果。
右上方
参数:该任务相关的参数。
保存:点击“保存”,系统将弹出下拉框让您选择保存的文件,保存相应的数据至数据中心。保存的内容包括“预处理结果”和“对接后结果”。其中“预处理结果”指经过蛋白预处理的结构;“对接后结果”指经过对接程序输出的结果。“对接后结果”保存范围有三个选项:勾选的行,仅保存结果表格中首列方框勾选的分子;筛选后的行,仅保存经过高级筛选后得到的分子(页面左上角显示的有效结果可以看到有多少分子通过了高级筛选);所有行,保存输出的所有结果。
下载:点击“下载”,系统将弹出下拉框让您选择下载的文件,下载相应的数据至本地。下载的内容包括“预处理结果”和“对接后结果”。其中“预处理结果”指经过蛋白预处理的结构;“对接后结果”指经过对接程序输出的结果。“对接后结果”下载范围有三个选项:勾选的行,仅下载结果表格中首列方框勾选的分子;筛选后的行,仅下载经过高级筛选后得到的分子(页面左上角显示的有效结果可以看到有多少分子通过了高级筛选);所有行,下载输出的所有结果。
右下方
3D 可视化界面,会根据左侧表格第一列的勾选状态进行显示;也可在下方 当前肽链 后的输入框中输入表格中的 ID 并回车显示具体分子,或者点击左右符号进行不同多肽的浏览。
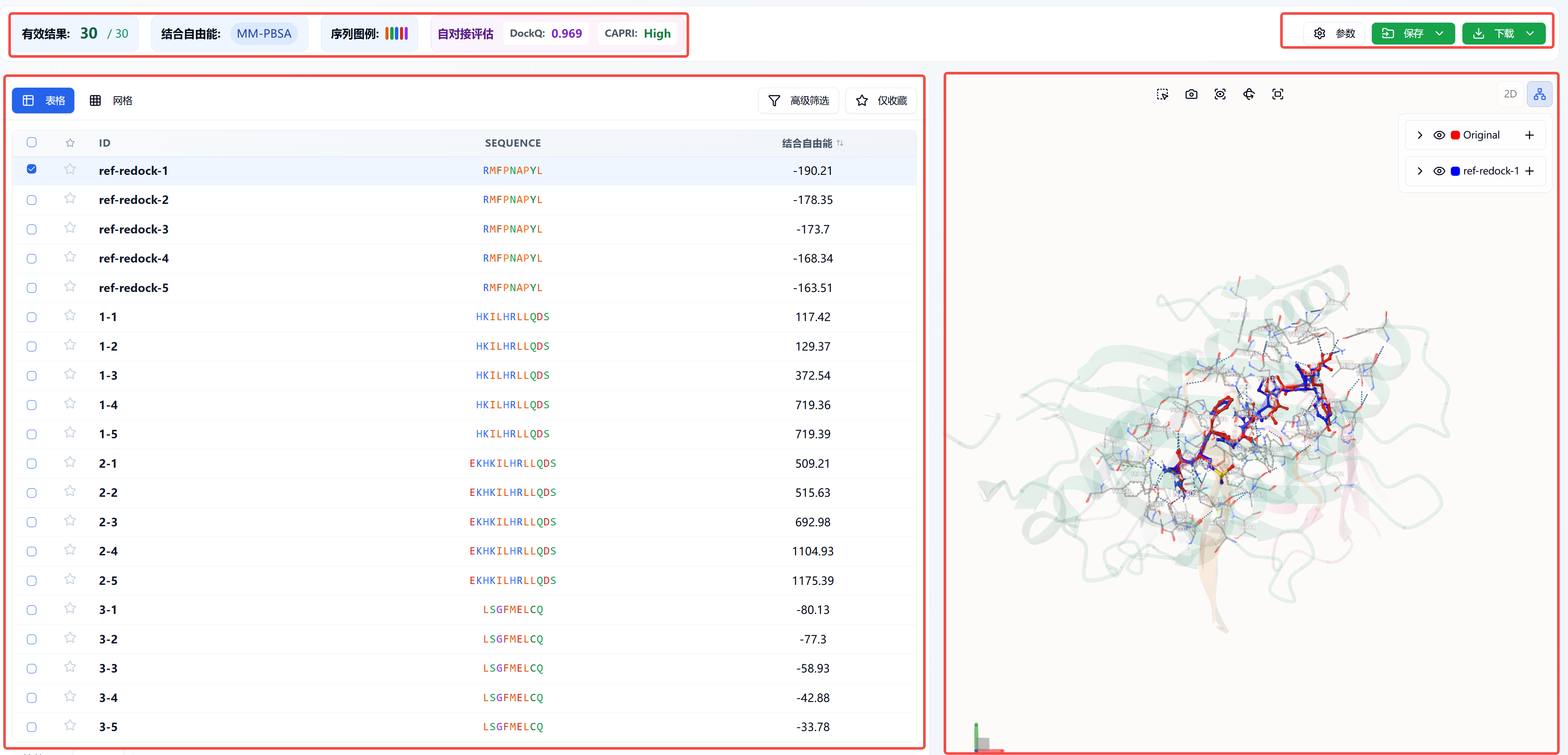
图 1. 计算结果页面
4. 相关文献
【1】Protein–peptide docking with a rational and accurate diffusion generative model, Nature Machine Intelligence, 2025, 7, 1308-1321.