Inno-PepDocking
1. Overview of Inno-PepDocking
Inno-PepDocking is a peptide docking module based on RAPiDock. This is a protein-peptide docking method utilizing a diffusion generative model, enabling rapid, accurate, and reasonable molecular docking at an all-atom level. On the RefPepDB-RecentSet benchmark, it achieves a Top-25 prediction success rate of 93.7%, which is 13.4% higher than AlphaFold2-Multimer, and offers approximately 270-fold speed improvement, with each complex taking only about 0.35 seconds. It supports all-atom modeling for 92 types of amino acid residues, including various post-translational modifications.
2. User Guide
(1) Protein Preprocessing
Protein Input
- Upload File: Upload .pdb files from your local machine.
- Data Center: Select .pdb files from the data center.
- Import from External Database: Enter a PDB ID to download the corresponding file from the PDB database.
Protein Preprocessing
Depending on the type of protein you upload, determine if preprocessing is needed. If the uploaded protein structure has already been preprocessed, you can proceed directly to the next step. If not, it is recommended to activate the preprocessing function.
Select Protein Chains to Retain: By default, all chains in the uploaded .pdb file are displayed and selected. Any unselected chain will not participate in subsequent computations.
Remove Water Molecules/Ligands: By default, all small molecules and water molecules within 5Å in the .pdb file are displayed. The system automatically removes water molecules outside 5Å, while you can decide to remove those within 5Å. Water molecules involved in water bridges are specially marked, and you can use the "Quickly remove non-bridging water molecules" button to remove them in one click. Any unselected ligands and their associated water molecules will be excluded from later calculations.
Protein Optimization Options
- Add Missing Residues/Repair Incorrect Structures: Optional, default is selected.
- Add Hydrogen Atoms: Optional, default is selected.
- Adjust Protonation State: Optional, default is selected, and pH is set to 7.4.
- Optimize Hydrogen Bond Network: Optional, default is selected.
- Energy Minimization: Optional, default is selected, and the force field is set to AMBER FF14SB.
AMBER FF14SB (Recommended): FF14SB is a protein force field parameter set in the AMBER package, used to describe atomic interactions in biomolecules. It is particularly suitable for protein systems, including extra parameters for side chain and key residue interactions within protein folding. AMBER FF14SB provides high accuracy and reliability in describing protein conformations and dynamics.
AMBER FF15IPQ: FF15IPQ is an improved protein force field in the AMBER package, offering greater accuracy and reliability than AMBER FF14IPQ. It includes more polarization effects and hydrogen bonding parameters, providing a more accurate description of protein electronic structure.
AMBER96: AMBER96 is an earlier version of the AMBER force field. While newer versions such as AMBER14 and AMBER18 exist, AMBER96 remains widely used in biomolecular simulations, especially in classic studies.
AMBER99SB: AMBER99SB is an improved version of the AMBER99 force field, with additional parameters to describe key interactions between protein side chains and within protein folding.
CHARMM36: CHARMM36 offers high precision and reliability for describing protein conformations and dynamics. It is widely used in protein and protein-ligand interaction studies and is one of the most commonly used force fields in biomolecular simulation.
(2) Peptide Input
- Upload File: Upload files in .fasta, .fas, .fa, or .txt format from your local machine.
- Data Center: Import fasta files from the data center.
- Text Input: Directly input peptide sequence text in the format: >sequence name\n sequence content. Multiple sequences are supported. Peptide input supports 20 standard amino acids and 72 non-standard amino acids as listed below: • 20 standard amino acids: G, A, V, L, I, P, F, Y, W, S, T, C, M, N, Q, D, E, K, R, H • 72 non-standard amino acids use the [XXX] format, e.g.: RMF[HYP]R[PTR]NAPYL HYP, SEP, TYS, ALY, TPO, PTR, DAL, MLE, M3L, DLE, DLY, AIB, MSE, DPR, MVA, NLE, MLY, SAR, ABA, FME, DAR, ORN, CGU, DPN, DTY, DTR, 4BF, DGL, DCY, MK8, MP8, GHP, ALC, BMT, MLZ, DVA, 3FG, DAS, 7ID, DSN, AR7, MEA, PHI, MAA, LPD, KCR, PCA, DGN, 2MR, DHI, ASA, MLU, YCP, DSG, DTH, OMY, FP9, DPP, HCS, SET, DBB, BTK, DAM, IIL, 3MY, SLL, PFF, HRG, DIL, DNE, MED, D0C
(3) Set Docking Parameters
Docking Method
Select a docking method. Currently, only RAPiDock is available.
Docking Center & Coordinate Settings
Two options are provided to define the docking center: select a peptide ligand in the complex or manually define the docking site. The default coordinates are automatically allocated by the system, and the default box length, width, and height represent the location and size of the binding pocket.
- Selecting a peptide ligand in the complex as the docking site: This option is only applicable if the complex contains peptide ligands. If there are multiple peptide ligands, the system shows the one with the largest molecular weight by default; the user can select other ligands from a dropdown. The geometric center (XYZ coordinates) and an appropriate box size will be calculated based on the peptide ligand's size.
- Custom docking site: In custom mode, you can click on a residue in the 3D view area, and the coordinate information for that residue will appear in the parameter panel. The system provides a default box size, but users should adjust the box size based on their understanding of the binding pocket. Too small a box may result in inaccurate results, while too large a box will increase computation time.
Number of Conformations
Set the maximum number of conformations to be output for each peptide ligand after docking.
Task Name
Task names can be user-defined or use the auto-assigned system default.
3. Result Analysis
The results page is shown in Figure 1. Top left:
- Valid Results: Number displayed in the current table / total number of results output.
- Binding Free Energy: Namely MM-PBSA. MM-PBSA is a popular method used in molecular simulations to calculate binding free energy. It is widely applied in studying interactions between proteins and ligands (drug molecules), proteins, or nucleic acids. A lower value indicates a more stable binding.
- Sequence Legend: Refers to the amino acid color codes in the SEQUENCE column below, divided into five categories: hydrophobic, polar, positively charged, negatively charged, and glycine.
- Self-docking Evaluation: When setting docking parameters, choosing the peptide ligand as the center will perform self-docking for that ligand, displaying the self-docking evaluation and corresponding results below. If multiple self-docking results are output, the value displayed is from the structure with the lowest MM-PBSA score (most stable binding).
Bottom left:
- ID: The output peptide's ID. The first part is the sequence index, the second is the conformation index for the same sequence.
- SEQUENCE: The input peptide sequence.
- Binding Free Energy: MM-PBSA, sortable by clicking the column; clicking a third time resets the order.
- Advanced Filtering: Filter results in the table below by binding free energy.
- Favorites Only: Show only favorited results in the table.
Top right:
Parameters: Parameters associated with the current task.
Save: Click "Save" to choose which files to save to the data center. The options are "Preprocessing Results" and "Post-docking Results". "Preprocessing Results" refers to structures after protein preprocessing; "Post-docking Results" refers to docking outputs. For "Post-docking Results", you can select: Checked rows (only the checked molecules in the first column of results table), Filtered rows (only the molecules which passed advanced filtering — you can see the number in the valid results section in the top left), or All rows (all output results).
Download: Click "Download" to choose which files to download. Contents include "Preprocessing Results" and "Post-docking Results", as above. For "Post-docking Results" you can select: Checked rows, Filtered rows, or All rows as described above.
Bottom right:
The 3D visualization interface displays the selected molecules according to the checkbox status in the first column of the left table. You can also enter an ID from the table in the field after "Current Peptide Chain" to show a specific molecule, or use the left/right arrows to browse different peptides.
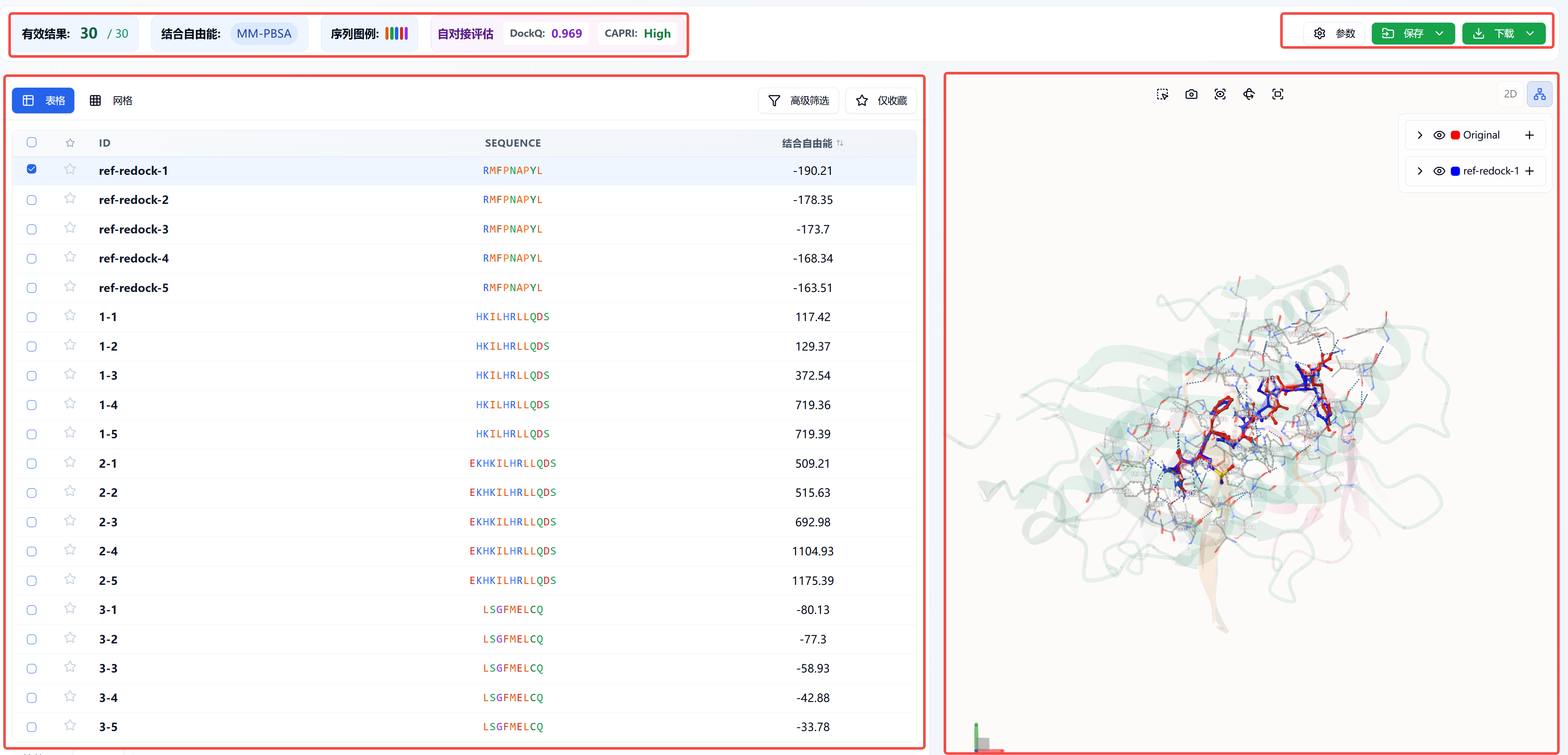
Figure 1. Results page
4. References
[1] Protein–peptide docking with a rational and accurate diffusion generative model, Nature Machine Intelligence, 2025, 7, 1308-1321.